
A kémia, és különösen a szerves kémia, rendkívül gazdag a molekulák háromdimenziós szerkezetének tanulmányozásában. A molekulák térbeli elrendezése, vagyis a konformáció, alapvetően befolyásolja fizikai és kémiai tulajdonságaikat, reakciókészségüket, sőt biológiai aktivitásukat is. Míg az izomerek olyan vegyületek, amelyek azonos összegképlettel rendelkeznek, de atomjaik kapcsolódási sorrendje vagy térbeli elrendezése eltér, addig a konformerek olyan térbeli elrendezések, amelyek egymásba átalakíthatók a kötések mentén történő rotációval anélkül, hogy kovalens kötések szakadnának fel vagy alakulnának ki. Ez a dinamikus természet teszi a konformációs analízist a modern kémia egyik központi területévé. A gyűrűs vegyületek esetében a konformációk még összetettebbé válnak, mivel a gyűrűs szerkezet korlátozza a rotáció szabadságát, és specifikus, gyakran feszültséggel járó elrendezéseket eredményez. Ezen a területen belül különösen izgalmas és sokoldalú a félszék konformáció vizsgálata, amely számos telítetlen gyűrűs vegyület, például a ciklohexén és bizonyos szénhidrátok alapvető szerkezeti egysége.
A konformációs analízis célja, hogy megértse és előre jelezze a molekulák preferált térbeli elrendezését, valamint az ezen elrendezések közötti energiakülönbségeket és az átalakulásukhoz szükséges energiaakadályokat. Ez a tudás kulcsfontosságú a reakciómechanizmusok magyarázatában, a gyógyszermolekulák tervezésében és az anyagtudományban is. A molekulák belső energiáját a kötések deformációja (kötéshossz és kötésszög eltérések az ideálistól), a torziós feszültség (a kötések mentén történő elfordulásból adódó feszültség) és a nem kötő atomok közötti van der Waals kölcsönhatások (sztérikus gátlás) határozzák meg. Egy adott konformáció stabilitását az határozza meg, hogy ezek a feszültségek milyen mértékben minimalizálódnak. A ciklohexán esetében a szék konformáció azért a legstabilabb, mert tökéletesen feszültségmentes, minimalizálva az összes szög- és torziós feszültséget. Azonban mi történik, ha egy kettős kötést vezetünk be a gyűrűbe? Ez a kérdés vezet el minket a félszék konformáció mélyebb megértéséhez.
A ciklohexán konformációi: alapok
Mielőtt mélyebben belemerülnénk a félszék konformáció rejtelmeibe, érdemes felidézni a ciklohexán, a hat tagú telített gyűrűs szénhidrogén konformációit. A ciklohexán a szerves kémia egyik leginkább tanulmányozott gyűrűs rendszere, és konformációs viselkedése alapvető referenciapontot jelent más gyűrűs rendszerek, így a ciklohexén félszék konformációjának megértéséhez is. A ciklohexán nem sík molekula, éppen azért, hogy minimalizálja a belső feszültségeket. Ha síkban lenne, akkor a C-C-C kötésszögek 120 fokosak lennének, ami jelentős szögfeszültséget okozna, mivel az ideális sp3 hibridizált szénatom kötésszöge 109,5 fok. Emellett a sík gyűrűben az összes hidrogénatom ekliptikus elrendezésben lenne, ami súlyos torziós feszültséget generálna.
A ciklohexán esetében a legstabilabb konformáció a szék konformáció. Ebben az elrendezésben az összes kötésszög közel ideális 109,5 fokos, és az összes szomszédos C-H kötés staggered (nyárs) elrendezésben van egymáshoz képest, ami minimalizálja a torziós feszültséget. A szék konformációban a hidrogénatomok kétféle pozíciót foglalhatnak el: axiális (a gyűrű síkjára merőlegesen, fel vagy le mutató) és ekvatoriális (a gyűrű síkjával párhuzamosan, kifelé mutató). Ezek a pozíciók a gyűrűátfordulás, vagy „ring flip” során egymásba átalakulnak, az axiális hidrogének ekvatoriálissá, az ekvatoriális hidrogének pedig axiálissá válnak.
A szék konformáció mellett léteznek kevésbé stabil konformációk is, mint például a kád konformáció és a csavart kád konformáció. A kád konformáció jelentős torziós feszültséggel jár, mivel négy hidrogénatom ekliptikus elrendezésben van, és a gyűrű „fenekén” lévő két hidrogénatom sztérikus gátlást (flagpole-flagpole interakció) is okoz. A csavart kád (twist-boat) konformáció egy átmeneti forma a szék és a kád között, amely valamivel stabilabb, mint a tiszta kád, mivel a torziós feszültségeket részben enyhíti a gyűrű enyhe elcsavarodása. Azonban mind a kád, mind a csavart kád konformáció jelentősen magasabb energiájú, mint a szék konformáció, ezért a ciklohexán szobahőmérsékleten szinte kizárólag szék konformációban található meg, gyorsan interkonvertálódva a két ekvivalens szék forma között.
A ciklohexán tanulmányozása alapvető fontosságú a gyűrűs rendszerek konformációs viselkedésének megértéséhez, mivel a szék konformáció a tökéletesen feszültségmentes, ideális elrendezést testesíti meg.
A gyűrűátfordulás egy dinamikus folyamat, amely során a szék konformációk egymásba alakulnak át egy csavart kád és egy kád átmeneti állapoton keresztül. Ez a folyamat szobahőmérsékleten gyorsan megy végbe, de alacsonyabb hőmérsékleten, például NMR spektroszkópia segítségével lelassítható és megfigyelhető. A szubsztituált ciklohexánok esetében a gyűrűátfordulás során a szubsztituensek preferáltan az ekvatoriális pozíciót foglalják el, mivel az axiális pozícióban jelentős sztérikus kölcsönhatásba léphetnek a gyűrű másik oldalán lévő axiális hidrogénekkel (1,3-diaxiális kölcsönhatás), ami destabilizálja a molekulát. Ez az elv alapvető fontosságú a konformációs analízisben, és segít megjósolni a gyűrűs molekulák preferált térbeli elrendezését.
A félszék konformáció születése: a ciklohexén
A ciklohexén bevezetése a konformációs analízisbe egy új dimenziót nyit meg. Míg a ciklohexánban minden szénatom sp3 hibridizált és tetraéderes geometriájú, addig a ciklohexénben két szénatom sp2 hibridizált, és egy kettős kötés részét képezi. Ez a kettős kötés radikálisan megváltoztatja a gyűrű geometriáját és konformációs lehetőségeit. Az sp2 hibridizált szénatomok és a hozzájuk kapcsolódó atomok egy síkban helyezkednek el, és a kötésszögük ideálisan 120 fok. Ez a sík rész beépül a hat tagú gyűrűbe, és jelentős hatással van a gyűrű egészének térbeli elrendezésére.
A ciklohexánnal ellentétben a ciklohexén nem képes tiszta szék vagy kád konformációt felvenni. Ennek oka éppen a kettős kötés merevsége és sík jellege. A szék konformációban a gyűrű „hullámos”, és a kötésszögek 109,5 fok körüliek. Ha egy kettős kötést helyeznénk ebbe a szerkezetbe, az sp2 szénatomok 120 fokos kötésszögei feszültséget okoznának, és a sík rész nem illeszkedne a gyűrű többi, nem sík részéhez. Hasonlóképpen, a kád konformáció is túl sok torziós feszültséget generálna az sp2 hibridizált szénatomok számára, amelyeknek síkban kell maradniuk.
Ehelyett a ciklohexén egy egyedi konformációt vesz fel, amelyet félszék konformációnak nevezünk. Ez az elnevezés jól tükrözi, hogy a konformáció a ciklohexán szék konformációjának egy „fél” részére emlékeztet, miközben alkalmazkodik a kettős kötés sík jellegéhez. A félszék konformáció a ciklohexén legstabilabb konformációja, mert ebben az elrendezésben minimalizálódnak a szög- és torziós feszültségek, amelyeket a kettős kötés bevezetése okoz.
A félszék konformáció kialakulásában kulcsszerepet játszik az sp2 hibridizáció. A kettős kötés két szénatomja és a hozzájuk kapcsolódó két hidrogénatom (vagy szubsztituens) egy síkban fekszik. Ez a négy atom egy merev, sík részt képez a gyűrűben. A gyűrű többi négy szénatomja (amelyek sp3 hibridizáltak) úgy rendeződik el, hogy a feszültségek a lehető legkisebbek legyenek. Az eredmény egy olyan szerkezet, ahol a kettős kötés síkjához képest a gyűrű „felülről” vagy „alulról” hajlik. Ez a hajlat hasonlít a ciklohexán szék konformációjának egyik felére, innen ered a neve.
A kettős kötés bevezetése a ciklohexán gyűrűjébe alapvetően megváltoztatja a konformációs lehetőségeket, és a félszék konformáció kialakulásához vezet, amely optimalizálja a feszültségeket a sík sp2 szénatomok és a hajlékony sp3 szénatomok között.
Ez a konformáció nem csupán elméleti érdekesség; a ciklohexén és származékai számos fontos kémiai reakcióban vesznek részt, és biológiailag aktív molekulák építőkövei is lehetnek. A félszék konformáció megértése elengedhetetlen a reakciók szelektivitásának és hozamának magyarázatához, valamint a szubsztituensek térbeli elhelyezkedésének előrejelzéséhez, ami közvetlenül befolyásolja a molekula kölcsönhatásait más molekulákkal.
A félszék konformáció részletes jellemzése
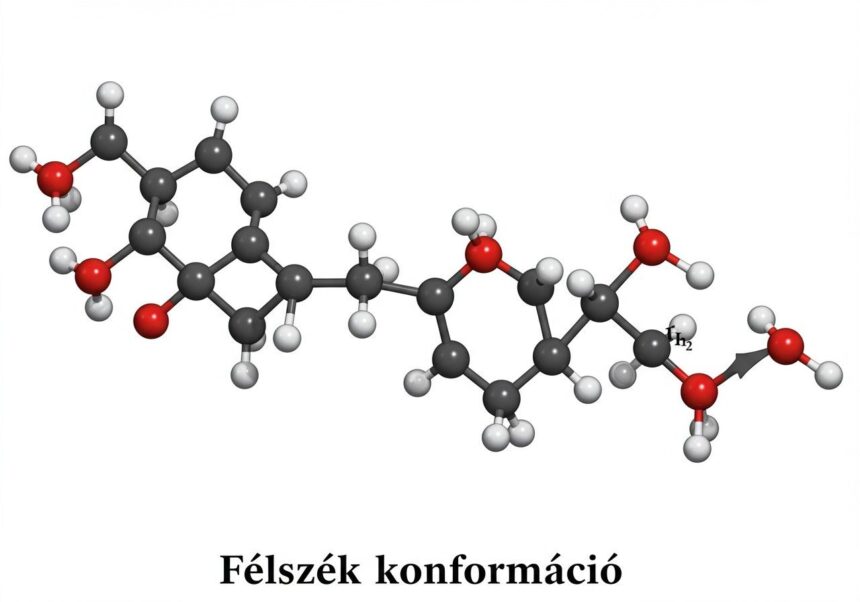
A félszék konformáció egyedi geometriai jellemzőkkel bír, amelyek megkülönböztetik más gyűrűs konformációktól. Ahogy a neve is sugallja, ez a konformáció a ciklohexán szék konformációjának egy részére emlékeztet, de a kettős kötés jelenléte miatt módosult. A ciklohexénben a C1=C2 kettős kötés és az ehhez kapcsolódó C3 és C6 szénatomok egy síkban helyezkednek el, vagyis a C1-C2-C3-C6-C1 fragmentum sík. A C3, C4, C5 és C6 szénatomok alkotják a gyűrű hajlított részét, amely a kettős kötés síkjához képest emelkedik vagy süllyed.
A félszék konformáció alapvető jellemzője a C2 szimmetria tengely. Ez a tengely áthalad a kettős kötés közepén, valamint a gyűrű szemközti oldalán lévő C4 szénatomon keresztül. Ez a szimmetria a molekula tükörképét önmagába viszi át, és segít megérteni a különböző atomok ekvivalenciáját vagy inekvivalenciáját a gyűrűben. A C1 és C2 szénatomok síkban vannak, és az ehhez a kettős kötéshez kapcsolódó hidrogének is síkban helyezkednek el. A C3 és C6 szénatomok, amelyek közvetlenül a kettős kötéshez kapcsolódnak, sp3 hibridizáltak, és enyhe torziós feszültséget viselnek, hogy illeszkedjenek a síkba, miközben a gyűrű többi részéhez is kapcsolódnak.
A kötésszögek tekintetében a C1=C2-C3 és C2=C1-C6 kötésszögek közel 120 fokosak, ami az sp2 hibridizációra jellemző. A C3-C4-C5 és C6-C5-C4 kötésszögek azonban közelebb állnak az sp3 ideális 109,5 fokos értékéhez, de a gyűrűs szerkezet miatt enyhe eltéréseket mutatnak. A torziós szögek is kulcsfontosságúak a feszültségek minimalizálásában. A C1-C2 kettős kötés torziós szöge természetesen 0 fok, mivel síkban van. A C2-C3-C4-C5 és C1-C6-C5-C4 dihedrális szögek azonban eltérnek a ciklohexán szék konformációjában tapasztalt ideális 60 fokos staggered elrendezéstől, alkalmazkodva a gyűrű feszült természetéhez.
A félszék konformációban a feszültségek minimalizálása összetett folyamat. A kettős kötés merevsége miatt a gyűrű nem tudja teljesen elkerülni a torziós feszültségeket, de a gyűrű hajlásával (puckering) a molekula megtalálja a legkevésbé feszült állapotot. Ezt gyakran az „envelope” (boríték) konformációval hozzák összefüggésbe, különösen öttagú gyűrűk esetében. A félszék konformáció tekinthető egy olyan boríték konformációnak, ahol a „boríték fedele” a kettős kötés síkja, és a gyűrű többi része, a C3, C4, C5, C6 atomok alkotják a „boríték testét”, amely ebből a síkból kiemelkedik. A C4 szénatom a leginkább kimozduló atom a kettős kötés síkjából.
Összehasonlítva a ciklohexán szék és kád konformációival, a félszék konformáció egy köztes állapotot képvisel a stabilitás és a feszültség tekintetében. Stabilabb, mint a tiszta kád konformáció, de gyakran tartalmaz némi feszültséget, amelyet a kettős kötés és a gyűrűs szerkezet kényszerít. Azonban ez a legstabilabb elérhető konformáció a ciklohexén számára, és a termodinamikai egyensúlyban domináns formát képviseli.
A félszék konformáció egy C2 szimmetriájú, feszültségoptimalizált elrendezés, ahol a kettős kötés síkja és a gyűrű hajlított része egyedülálló módon ötvöződik, biztosítva a ciklohexén és rokon vegyületek stabilitását.
A félszék konformációban az atomok térbeli elhelyezkedése alapvető fontosságú a szubsztituensek pozícióinak és a reakciók szelektivitásának megértésében. Míg a ciklohexánban egyértelműen beszélhetünk axiális és ekvatoriális pozíciókról, a félszék konformációban ezek a fogalmak módosulnak, és pszeudo-axiális és pszeudo-ekvatoriális pozíciókról van szó, amelyek a későbbiekben kerülnek részletezésre. Ez a finom különbség kulcsfontosságú a gyűrűs telítetlen vegyületek kémiai viselkedésének pontos leírásában.
Energetika és dinamika

A molekulák konformációi nem statikusak; folyamatosan átalakulnak egymásba, különösen szobahőmérsékleten, ahol elegendő energia áll rendelkezésre az energiagátak leküzdéséhez. A ciklohexén félszék konformációjának dinamikája egy különösen érdekes terület, amely magában foglalja a különböző félszék formák közötti átmeneteket és az ezekhez kapcsolódó energiaprofilokat.
A ciklohexén félszék konformációja, bár a legstabilabb forma, nem egyetlen merev szerkezet. Két ekvivalens félszék konformáció létezik, amelyek egymás tükörképei (enantiomerek, ha a gyűrű szubsztituált). Ezek az enantiomer félszék formák pszeudorotáció (álrotáció) révén alakulnak át egymásba. A pszeudorotáció egy olyan folyamat, amely során a gyűrű alakja deformálódik, de nem történik meg a teljes gyűrűátfordulás, mint a ciklohexán esetében. Ehelyett a gyűrű hajlított része „hullámzik”, és a kettős kötés síkjához képest a gyűrű felülről vagy alulról történő hajlása megváltozik.
A pszeudorotáció során a C4 szénatom, amely a kettős kötés síkjából a leginkább kimozduló atom, átlépi a síkot, és a gyűrű hajlása megfordul. Ez a folyamat viszonylag alacsony energiaakadállyal rendelkezik, ami azt jelenti, hogy szobahőmérsékleten nagyon gyorsan megy végbe. Az aktiválási energia ehhez az interkonverzióhoz jellemzően 5-10 kcal/mol (20-40 kJ/mol) nagyságrendű, ami jelentősen alacsonyabb, mint a ciklohexán gyűrűátfordulásának energiagátja (kb. 10-11 kcal/mol). Ez a különbség abból adódik, hogy a ciklohexénben a kettős kötés merevsége miatt kevesebb mozgásszabadság áll rendelkezésre, és a gyűrű „puhább”, könnyebben deformálódik.
Az energiaprofil vizsgálata során látható, hogy a két ekvivalens félszék konformáció közötti átmenet egy magasabb energiájú átmeneti állapoton keresztül történik. Ez az átmeneti állapot gyakran egy „síkszerű” vagy „lapos” félszék formához hasonlítható, ahol a gyűrű hajlása minimális. Azonban fontos megjegyezni, hogy a gyűrű sosem válik teljesen síkba, mert az rendkívül magas szög- és torziós feszültséggel járna. Ehelyett az átmeneti állapot egy olyan konformáció, ahol a feszültségek átmenetileg magasabbak, de még mindig minimalizálva vannak a kettős kötés síkjának fenntartása mellett.
A félszék konformáció dinamikus jellege fontos következményekkel jár a szubsztituált ciklohexének kémiai viselkedésére. Ha egy szubsztituens van jelen a gyűrűn, akkor a két félszék konformáció már nem feltétlenül ekvivalens energetikailag. A szubsztituens sztérikus kölcsönhatásai a gyűrű többi részével befolyásolják, hogy melyik félszék forma lesz a preferált. Például, ha egy nagy szubsztituens van jelen, akkor az a konformáció lesz stabilabb, amelyben a szubsztituens a legkevésbé gátolt pozíciót foglalja el. Ez az elv alapvető a reakciók sztereokémiájának megértésében, mivel a reaktáns molekula preferált konformációja befolyásolhatja, hogy melyik termék képződik nagyobb mennyiségben.
A különböző konformációk közötti energiakülönbségeket és az aktiválási energiákat számos kísérleti technikával lehet vizsgálni, többek között NMR spektroszkópiával és számítógépes kémiai módszerekkel. Az NMR spektroszkópia, különösen alacsony hőmérsékleten, lehetővé teszi a gyorsan interkonvertálódó konformerek „befagyasztását”, és ezáltal az egyes formák spektrumának megfigyelését. A számítógépes kémia, például a kvantumkémiai számítások, pontosan meg tudják becsülni a konformerek energiáit és az átmeneti állapotok energiagátjait, kiegészítve a kísérleti adatokat.
A ciklohexén félszék konformációja dinamikus egyensúlyban létezik két ekvivalens forma között, amelyek pszeudorotációval alakulnak át egymásba, viszonylag alacsony energiaakadályon keresztül.
Ez a dinamikus viselkedés nem csupán elméleti érdekesség, hanem gyakorlati jelentőséggel is bír. Például a gyógyszertervezésben, ahol a molekula térbeli alakja alapvető a receptorokhoz való kötődés szempontjából, a konformációk közötti gyors átmenetek megértése kulcsfontosságú lehet. Egy gyógyszermolekula rugalmassága és a preferált konformációinak ismerete segíthet optimalizálni a kötődést és a biológiai aktivitást.
Szubsztituensek hatása a félszék konformációra
A szubsztituensek bevezetése a gyűrűs molekulákba jelentősen befolyásolja azok konformációs viselkedését. A ciklohexánban a szubsztituensek preferáltan az ekvatoriális pozíciót foglalják el, hogy minimalizálják az 1,3-diaxiális kölcsönhatásokat. A félszék konformációval rendelkező ciklohexének esetében azonban a helyzet kissé eltérő, mivel a gyűrű geometriája és a kettős kötés jelenléte módosítja a lehetséges pozíciókat és a sztérikus kölcsönhatásokat.
A félszék konformációban, a C1=C2 kettős kötés síkja miatt, már nem beszélhetünk tiszta axiális és ekvatoriális pozíciókról a kettős kötéshez közvetlenül kapcsolódó szénatomok (C3 és C6) hidrogénjei vagy szubsztituensei esetében. Ehelyett a pszeudo-axiális és pszeudo-ekvatoriális fogalmakat használjuk. Ezek a pozíciók a ciklohexán axiális és ekvatoriális pozícióihoz hasonlítanak, de a gyűrű deformációja miatt nem pontosan merőlegesek vagy párhuzamosak a gyűrű síkjával.
Tekintsük a C3 (vagy C6) szénatomot. Ennek a szénatomnak két hidrogénje (vagy szubsztituense) van. Az egyik a kettős kötés síkjához képest feljebb vagy lejjebb helyezkedik el, és közelít az axiális pozícióhoz – ezt nevezzük pszeudo-axiálisnak. A másik hidrogén (vagy szubsztituens) pedig inkább a gyűrű síkjával párhuzamosan, kifelé mutató pozíciót foglal el – ezt nevezzük pszeudo-ekvatoriálisnak. A C4 és C5 szénatomok esetében, amelyek távolabb vannak a kettős kötéstől és inkább „szék-szerű” környezetben vannak, a pozíciók jobban hasonlítanak a ciklohexán axiális és ekvatoriális pozícióihoz, de még itt is van némi torzítás a gyűrű feszültsége miatt.
A szubsztituensek preferenciája a pszeudo-ekvatoriális pozíció iránt a ciklohexénekben is megfigyelhető, hasonlóan a ciklohexánhoz. A nagyobb térigényű szubsztituensek (pl. metil, terc-butil csoportok) általában a pszeudo-ekvatoriális pozíciót részesítik előnyben, hogy minimalizálják a sztérikus gátlást más hidrogénekkel vagy szubsztituensekkel. A pszeudo-axiális pozícióban lévő szubsztituensek közelebb kerülhetnek a gyűrű más részeihez, ami destabilizáló kölcsönhatásokat eredményezhet.
Például, egy 3-metilciklohexén esetében a metilcsoport a C3 szénatomon lehet pszeudo-axiális vagy pszeudo-ekvatoriális pozícióban. A pszeudo-ekvatoriális izomer lesz a stabilabb, mivel a metilcsoport kevésbé gátolt ebben az elrendezésben. Azonban a különbség a két konformer energiája között általában kisebb, mint a ciklohexánban, mivel a félszék konformáció gyűrűje rugalmasabb és jobban el tudja viselni a sztérikus feszültségeket.
| Pozíció típusa | Leírás | Preferált szubsztituens |
|---|---|---|
| Pszeudo-axiális | A gyűrű síkjára merőlegesen, fel vagy le mutató irányba esik, de a gyűrű deformációja miatt nem pontosan axiális. | Kisebb méretű, hidrogénhez hasonló szubsztituensek. |
| Pszeudo-ekvatoriális | A gyűrű síkjával nagyjából párhuzamosan, kifelé mutató irányba esik, de a gyűrű deformációja miatt nem pontosan ekvatoriális. | Nagyobb méretű szubsztituensek a sztérikus gátlás minimalizálása érdekében. |
A szubsztituensek elhelyezkedése nem csak a stabilitást, hanem a reakciók szelektivitását is befolyásolja. Például, egy ciklohexén gyűrűn lejátszódó addíciós reakciók (pl. epoxidáció, hidrogénezés) sztereokémiája nagymértékben függ attól, hogy a reagens melyik oldalról tudja megközelíteni a kettős kötést. Ha egy nagy szubsztituens van a gyűrű egyik oldalán, az gátolhatja a reagens hozzáférését arról az oldalról, elősegítve a reakciót a kevésbé gátolt oldalról. Ez a jelenség a sztereoszelektivitás alapja, és kulcsfontosságú a szerves szintézisben.
A szubsztituensek a félszék konformációban pszeudo-axiális és pszeudo-ekvatoriális pozíciókat foglalnak el, ahol a nagyobb csoportok a pszeudo-ekvatoriális elrendezést részesítik előnyben a sztérikus kölcsönhatások minimalizálása érdekében, ami befolyásolja a molekula stabilitását és reakciókészségét.
A szubsztituensek hatásának pontos megértése elengedhetetlen a komplex biológiai molekulák, például a szteroidok vagy a terpének konformációjának és aktivitásának magyarázatában, amelyek gyakran tartalmaznak telítetlen gyűrűket. A gyógyszertervezés során a hatóanyagok térbeli elrendezésének optimalizálása, a szubsztituensek gondos megválasztása és pozícionálása alapvető fontosságú a kívánt biológiai válasz eléréséhez.
Félszék konformáció más gyűrűs rendszerekben
Bár a ciklohexén a félszék konformáció prototípusa, ez az elrendezés nem korlátozódik kizárólag hat tagú, egy kettős kötést tartalmazó gyűrűkre. A félszék konformáció, vagy ahhoz nagyon hasonló, feszültségoptimalizált gyűrűgeometriák számos más ciklusos vegyületben is megjelennek, különösen szénhidrátokban és egyéb heterociklusos rendszerekben. Ezek a molekulák gyakran kulcsfontosságúak a biológiai folyamatokban, és a konformációjuk alapvetően befolyásolja biológiai aktivitásukat.
Szénhidrátok: a furanóz gyűrűk
A szénhidrátok, mint például a cukrok, rendkívül sokoldalú molekulák, amelyek gyakran gyűrűs formában léteznek. Az öttagú cukorgyűrűket, mint például a ribóz és a dezoxiribóz (amelyek a DNS és RNS gerincét alkotják), furanóz gyűrűknek nevezzük. Ezek a furanóz gyűrűk nem síkban helyezkednek el, hanem különféle „puckering” (gyűrűhajlás) konformációkat vesznek fel, hogy minimalizálják a torziós és szögfeszültségeket. A furanóz gyűrűk leggyakoribb konformációi az envelope (boríték) és a félszék (half-chair) konformációk.
Az envelope konformációban a gyűrű öt atomjából négy egy síkban helyezkedik el, míg az ötödik atom kilép ebből a síkból, mint egy boríték fedele. Például, a ribóz esetében a C2′-endo vagy C3′-endo envelope konformációk gyakoriak, attól függően, hogy a C2′ vagy a C3′ szénatom mozdul el a síkból. A félszék konformáció a furanóz gyűrűk esetében hasonló a ciklohexén félszékéhez, de öttagú gyűrűben. Itt két szomszédos atom és a hozzájuk kapcsolódó atomok egy síkban helyezkednek el, míg a gyűrű többi része hajlított. A furanóz gyűrűk esetében a félszék és envelope konformációk közötti energiakülönbségek gyakran nagyon kicsik, és gyorsan átalakulnak egymásba.
A furanóz gyűrűk konformációja kritikus jelentőségű a nukleinsavak (DNS és RNS) szerkezetében és működésében. A ribózgyűrű puckeringje befolyásolja a bázisok térbeli elrendezését, és ezáltal az egész kettős spirál vagy az RNS szerkezetét. Például a DNS-ben a dezoxiribóz gyűrűk leggyakrabban C2′-endo envelope konformációban vannak, míg az RNS-ben a ribóz gyűrűk gyakrabban C3′-endo envelope konformációt vesznek fel. Ezek a finom különbségek alapvetőek a DNS és RNS eltérő szerkezeti rugalmasságában és biológiai funkcióiban.
A puckering paraméterek (például a pseudorotation fázisszöge és az amplitúdója) egy matematikai módszert biztosítanak a furanóz gyűrűk konformációjának pontos leírására. Ezek a paraméterek lehetővé teszik a kutatók számára, hogy kvantitatívan jellemezzék a gyűrű hajlását, és nyomon kövessék a konformációs változásokat különböző körülmények között vagy molekuláris kölcsönhatások során.
Más cikloalkének és heterociklusok
A félszék konformáció elve kiterjeszthető más méretű telítetlen gyűrűkre is. Például a cikloheptén (hét tagú gyűrű egy kettős kötéssel) és a ciklooktén (nyolc tagú gyűrű egy kettős kötéssel) is felvesznek preferált, nem sík konformációkat, amelyek minimalizálják a feszültségeket. Ezek a konformációk gyakran bonyolultabbak, mint a ciklohexén félszék formája, és több lehetséges stabil konformert is magukban foglalhatnak, mint például a csavart szék, csavart kád, vagy akár „korona” alakú formák, de az alapelv, miszerint a kettős kötés síkja kényszeríti a gyűrű többi részét egy specifikus hajlásra, továbbra is érvényes.
A heterociklusos vegyületek, amelyek a gyűrűben szénatomok mellett más atomokat (pl. oxigén, nitrogén, kén) is tartalmaznak, szintén felvehetnek félszék-szerű konformációkat, ha kettős kötés van jelen a gyűrűben. Például a dihidropirán, amely egy hat tagú gyűrű egy oxigénatommal és egy kettős kötéssel, szintén félszék konformációt vesz fel. Itt az oxigénatom bevezetése módosíthatja a kötésszögeket és a torziós feszültségeket, de az alapvető félszék geometria megmarad, mint a legstabilabb elrendezés.
Az olyan molekulák, mint a kumarinok vagy a krománok (amelyek gyakoriak a növényi eredetű vegyületek között), szintén tartalmazhatnak telítetlen heterociklusos gyűrűket, amelyek konformációja jelentősen befolyásolja biológiai aktivitásukat. A gyűrűs feszültségek, a kettős kötések és a heteroatomok kölcsönhatása együttesen határozza meg a preferált térbeli elrendezést, amely gyakran félszék-szerű jellemzőket mutat.
A félszék konformáció elve széles körben alkalmazható, a ciklohexénen túl is, különösen a biológiailag fontos furanóz gyűrűkben, valamint más telítetlen cikloalkénekben és heterociklusos rendszerekben, ahol a kettős kötés síkja kényszeríti a gyűrűt egy specifikus, feszültségoptimalizált hajlásra.
A különböző gyűrűs rendszerek konformációinak megértése nemcsak a szerkezeti kémia szempontjából fontos, hanem a gyógyszerkutatásban is kulcsfontosságú. A gyógyszermolekulák és a biológiai célpontjaik (pl. enzimek, receptorok) közötti kölcsönhatások rendkívül érzékenyek a molekulák térbeli alakjára. Egy molekula preferált konformációjának ismerete lehetővé teszi a kutatók számára, hogy racionálisan tervezzenek új gyógyszereket, amelyek optimális kötődést mutatnak a célpontjukhoz, ezáltal növelve hatékonyságukat és szelektivitásukat.
A félszék konformáció azonosítása és vizsgálata
A félszék konformáció, mint bármely más molekuláris szerkezet, nem csupán elméleti konstrukció. Különféle kísérleti és számítási módszerekkel azonosítható és jellemezhető. Ezek a technikák lehetővé teszik a kémikusok számára, hogy megerősítsék a konformáció létezését, meghatározzák a pontos geometriai paramétereit, és tanulmányozzák a dinamikus viselkedését, például az interkonverziós energiagátakat.
NMR spektroszkópia
A nukleáris mágneses rezonancia (NMR) spektroszkópia az egyik legerősebb eszköz a molekulák szerkezetének és dinamikájának vizsgálatára oldatban. Az NMR képes információt szolgáltatni az atomok kapcsolódási sorrendjéről, a kémiai környezetükről és a térbeli elrendezésükről. A félszék konformáció vizsgálatában különösen hasznosak a J-csatolási állandók és a kémiai eltolódások.
A vicinális protonok közötti J-csatolási állandók (azaz három kötésen keresztül kapcsolódó protonok közötti csatolás) erősen függnek a dihedrális szögtől (Karplus-reláció). A ciklohexén félszék konformációjában a különböző protonok közötti dihedrális szögek egyedi mintázatot mutatnak, amelyek alapján megkülönböztethető más konformációktól. Például a C3 és C6 szénatomokon lévő pszeudo-axiális és pszeudo-ekvatoriális protonok eltérő J-csatolási állandókat mutatnak egymással és a C4, C5 protonjaival. Ezeknek az értékeknek az elemzése lehetővé teszi a gyűrű hajlásának és a szubsztituensek preferált pozíciójának meghatározását.
Az alacsony hőmérsékletű NMR spektroszkópia különösen értékes a dinamikus konformációs folyamatok vizsgálatában. Ha a hőmérsékletet annyira lecsökkentjük, hogy a konformerek közötti interkonverzió lelassul, az NMR spektrumban az egyes konformerekre jellemző jelek külön-külön megjelenhetnek. Ezáltal közvetlenül megfigyelhetők a különböző félszék formák, és az energiagát a pszeudorotációhoz kiszámítható a koaleszcencia hőmérsékletéből.
Röntgenkrisztallográfia
A röntgenkrisztallográfia a legközvetlenebb módszer a molekulák háromdimenziós szerkezetének meghatározására szilárd fázisban. Amikor egy vegyület kristályos formában van, a röntgensugarak diffrakciós mintázata alapján pontosan meghatározható az atomok térbeli elhelyezkedése, a kötéshosszok, a kötésszögek és a torziós szögek. Ezáltal a félszék konformáció pontos geometriai paraméterei közvetlenül mérhetők.
A röntgenkrisztallográfiai adatok megerősítik a félszék konformáció létezését, és részletes információt szolgáltatnak a gyűrű hajlásáról, a kettős kötés síkjáról, és arról, hogy mely atomok mozdulnak el a leginkább a síkból. Ez különösen hasznos lehet a szubsztituált ciklohexének vagy más komplex, félszék konformációval rendelkező molekulák, például szénhidrátok kristályszerkezetének vizsgálatában.
Számítógépes kémia
A számítógépes kémia, különösen a kvantumkémiai számítások és a molekuláris mechanika módszerei, nélkülözhetetlen eszközök a konformációs analízisben. Ezek a módszerek lehetővé teszik a molekulák energiájának és geometriájának elméleti becslését, valamint a potenciális energiagörbék feltérképezését a konformációs változások során.
- Molekuláris mechanika (MM): Ez a módszer az atomokat rugókkal összekapcsolt golyóknak tekinti, és empirikus paraméterek (kötéshossz, kötésszög, torziós szögek, van der Waals erők) alapján számítja ki a molekula energiáját. Gyorsan képes feltérképezni a potenciális energiafelületet, és megtalálni a különböző konformációs minimumokat és átmeneti állapotokat. Ideális a nagy molekulák konformációs kereséséhez.
- Kvantumkémiai számítások (pl. DFT, ab initio): Ezek a módszerek az elektronok viselkedését írják le kvantummechanikai elvek alapján, és sokkal pontosabb energiákat és geometriákat szolgáltatnak, mint a molekuláris mechanika. Képesek pontosan meghatározni a félszék konformáció geometriáját, a különböző konformerek közötti energiakülönbségeket és az átmeneti állapotok aktiválási energiáit. Bár számításigényesebbek, megbízható referenciaként szolgálnak a kísérleti adatok értelmezéséhez.
A számítógépes kémia különösen hasznos a konformációs energiaprofilok kidolgozásában, a pszeudorotációs útvonalak azonosításában és a szubsztituensek konformációra gyakorolt hatásának előrejelzésében. Segítségével olyan molekulákat is vizsgálhatunk, amelyeket kísérletileg nehéz vagy lehetetlen tanulmányozni.
Az NMR spektroszkópia, a röntgenkrisztallográfia és a számítógépes kémia együttesen biztosítják a félszék konformáció mélyreható megértését, lehetővé téve a geometriai jellemzők, a dinamikus viselkedés és a szubsztituensek hatásának pontos elemzését.
Ezek a vizsgálati módszerek nemcsak a félszék konformáció alapkutatásában fontosak, hanem a gyakorlati alkalmazásokban is, például a gyógyszerfejlesztésben, ahol a molekulák térbeli szerkezetének pontos ismerete elengedhetetlen a hatóanyagok tervezéséhez és optimalizálásához. Az adatok összehasonlítása és a különböző módszerek kiegészítő jellege biztosítja a legátfogóbb képet a molekulák konformációs viselkedéséről.
Biológiai és gyógyszerészeti jelentőség
A félszék konformáció és általában a molekulák konformációs viselkedésének megértése messze túlmutat a puszta akadémiai érdekességen. Különösen a biológiai rendszerekben és a gyógyszerfejlesztésben van alapvető jelentősége, mivel a molekulák térbeli alakja kritikus tényező a biológiai folyamatokban és a gyógyszerek hatásmechanizmusában.
Enzim-szubsztrát kölcsönhatások
A biológiai rendszerekben az enzimek katalizálják a legtöbb kémiai reakciót, és működésük kulcsfontosságú a sejtek életben maradásához. Az enzimek rendkívül szelektívek, ami azt jelenti, hogy általában csak egy specifikus szubsztrátot vagy szubsztrátcsoportot alakítanak át. Ez a szelektivitás a szubsztrát molekula és az enzim aktív centruma közötti komplementer térbeli illeszkedésen alapul, amelyet gyakran a „kulcs-zár” mechanizmussal magyarázunk. Ha egy szubsztrát molekula tartalmaz egy félszék konformációt (például egy telítetlen cukorgyűrűt), akkor annak pontos térbeli elrendezése alapvető fontosságú lesz ahhoz, hogy az enzim aktív centrumába illeszkedjen.
Például, számos szénhidrát-átalakító enzim működésében a furanóz gyűrűk konformációja (félszék vagy envelope) kulcsszerepet játszik. Az enzim felismeri és megköti a szubsztrát specifikus konformációját, és gyakran a reakció során maga a gyűrű konformációja is megváltozik, hogy elősegítse a katalitikus folyamatot. Egy olyan szubsztrát, amely nem képes felvenni a megfelelő konformációt, vagy amelynek preferált konformációja eltér az enzim által felismerttől, nem lesz hatékonyan átalakítva.
Gyógyszertervezés: konformáció-aktivitás összefüggések
A gyógyszertervezés során a molekulák konformációjának megértése az egyik legfontosabb szempont. Egy gyógyszermolekula biológiai hatását alapvetően befolyásolja, hogy milyen térbeli alakot vesz fel, és hogyan lép kölcsönhatásba a célfehérjékkel (receptorok, enzimek, ioncsatornák). A félszék konformációval rendelkező molekulák, mint például bizonyos szteroidok, terpének vagy nukleozid analógok, esetében a gyűrű hajlása és a szubsztituensek pszeudo-axiális/pszeudo-ekvatoriális elhelyezkedése döntő lehet a hatóanyag kötődési affinitása és szelektivitása szempontjából.
A gyógyszertervezők gyakran használnak konformációs analízist és molekuláris modellezést, hogy előre jelezzék a potenciális gyógyszermolekulák preferált konformációit. Céljuk, hogy olyan molekulákat szintetizáljanak, amelyek a célreceptorhoz optimálisan illeszkedő konformációt vesznek fel. Ha egy molekula túl merev, és nem tud alkalmazkodni a receptorhoz, vagy ha a preferált konformációja nem illeszkedik, akkor valószínűleg nem lesz hatékony gyógyszer. Ezzel szemben, ha egy molekula túl rugalmas, és sokféle konformációt képes felvenni, az „elvesztegetett” energiát jelenthet, mivel csak egy vagy néhány konformáció lesz biológiailag aktív.
A konformációs flexibilitás és a preferált konformációk közötti finom egyensúly megértése kulcsfontosságú a gyógyszerhatóanyagok optimalizálásában. A félszék konformációval rendelkező gyűrűk inherent rugalmassága és a pszeudorotációs képességük lehetővé teszi számukra, hogy bizonyos mértékig alkalmazkodjanak a biológiai környezethez, de ugyanakkor megőrizzenek egy jellegzetes térbeli alakot, ami a szelektivitásuk alapja lehet.
Nukleinsavak működése
Ahogy korábban említettük, a furanóz gyűrűk konformációja (amelyek gyakran félszék vagy envelope formákat vesznek fel) alapvető a DNS és RNS szerkezetében. A nukleotidok és a nukleozidok ribóz vagy dezoxiribóz egységeinek puckeringje befolyásolja a bázisok egymáshoz viszonyított térbeli elhelyezkedését, ami közvetlenül hat a kettős spirál stabilitására, a fehérjékkel való kölcsönhatásokra és az RNS hajtogatására.
A dezoxiribóz C2′-endo félszék konformációja a DNS B-formjában, míg a ribóz C3′-endo félszék konformációja az RNS A-formjában kulcsfontosságú. Ezek a konformációs különbségek határozzák meg a DNS és RNS eltérő szerkezeti merevségét, a bázisok közötti távolságot és a foszfátgerinc térbeli elrendezését. Bármilyen változás ezekben a konformációkban befolyásolhatja a génexpressziót, a replikációt, a transzkripciót és a fehérjeszintézist.
A félszék konformáció biológiai jelentősége elengedhetetlen az enzim-szubsztrát illeszkedésben, a gyógyszerek célponthoz való kötődésében és a nukleinsavak komplex szerkezetének fenntartásában, bemutatva a molekuláris geometria mélyreható hatását az élő rendszerek működésére.
Összességében a félszék konformáció tanulmányozása nem csupán egy kémiai elmélet, hanem egy olyan alapvető eszköz, amely segít megérteni a molekulák viselkedését a legkomplexebb biológiai rendszerekben is. A konformációs analízis révén szerzett ismeretek lehetővé teszik a tudósok számára, hogy manipulálják a molekulák térbeli alakját, és ezáltal új gyógyszereket, anyagokat és biológiai eszközöket fejlesszenek ki.
A konformáció dinamikus természete és a kutatás kihívásai
A molekulák konformációja nem egy statikus állapot, hanem egy dinamikus egyensúlyban lévő halmaz, amely folyamatosan változik. A félszék konformáció és más gyűrűs rendszerek viselkedésének mélyreható megértése számos kihívást és kutatási lehetőséget rejt magában. A dinamikus folyamatok, a környezeti tényezők hatása és az elméleti modellek pontossága mind hozzájárulnak a konformációs analízis komplexitásához és izgalmához.
Gyors egyensúlyok és átmeneti állapotok
A molekulák konformációi, mint láttuk, gyorsan interkonvertálódnak egymásba. A félszék konformáció esetében a pszeudorotáció viszonylag alacsony energiaakadállyal rendelkezik, ami azt jelenti, hogy szobahőmérsékleten a két ekvivalens félszék forma rendkívül gyorsan átalakul egymásba. Ez a gyors egyensúly kihívást jelenthet a kísérleti megfigyelés szempontjából, mivel a legtöbb spektroszkópiai módszer csak az átlagolt szerkezetet látja. Azonban az alacsony hőmérsékletű NMR technikák lehetővé teszik az egyes konformerek „befagyasztását”, és ezáltal az egyedi spektrumok megfigyelését, ami kritikus információt szolgáltat az energiagátakról és a konformerek arányáról.
Az átmeneti állapotok, amelyek a konformerek közötti energiagörbe csúcsán helyezkednek el, rendkívül rövid életűek és közvetlenül nem figyelhetők meg kísérletileg. Azonban a számítógépes kémiai módszerek, különösen a kvantumkémiai számítások, képesek modellezni ezeket az átmeneti állapotokat, meghatározni a geometriájukat és az energiájukat. Ez az információ elengedhetetlen a reakciómechanizmusok és a konformációs változások kinetikájának teljes megértéséhez.
Környezeti tényezők (oldószer, hőmérséklet) hatása
A molekulák konformációja nem csak a belső kötések és a sztérikus kölcsönhatások függvénye, hanem jelentősen befolyásolják a környezeti tényezők is. Az oldószer polaritása, ionos ereje vagy hidrogénkötés-donor/akceptor képessége mind hatással lehet a molekula preferált konformációjára. Például, egy poláris oldószer stabilizálhatja azokat a konformációkat, amelyekben a dipólusmomentum nagyobb, vagy amelyekben jobban ki vannak téve a poláris csoportok az oldószernek.
A hőmérséklet szintén kulcsfontosságú tényező. Magasabb hőmérsékleten a molekulák nagyobb kinetikus energiával rendelkeznek, ami lehetővé teszi számukra, hogy könnyebben leküzdjék az energiagátakat, és így szélesebb konformációs tartományt fedezzenek fel. Ez azt jelenti, hogy a kevésbé stabil, magasabb energiájú konformerek aránya megnőhet magasabb hőmérsékleten. Az alacsony hőmérséklet ezzel szemben „befagyasztja” a molekulát a legstabilabb konformációjába vagy a legmélyebb energiagátakkal elválasztott konformerekbe.
Ezeknek a külső hatásoknak a pontos megértése elengedhetetlen a biológiai rendszerekben zajló folyamatok modellezéséhez, ahol a molekulák komplex és változatos környezetben működnek. Egy gyógyszermolekula konformációja egy oldatban eltérhet attól, amit egy receptor kötőhelyén felvesz, és ezeket a különbségeket figyelembe kell venni a tervezés során.
Elméleti és kísérleti eredmények összehasonlítása
A modern konformációs analízis a kísérleti és elméleti módszerek szoros együttműködésére épül. Az elméleti számítások (kvantumkémia, molekuláris mechanika) képesek előre jelezni a lehetséges konformációkat, azok energiáit és az átmeneti állapotokat. A kísérleti módszerek (NMR, röntgenkrisztallográfia) pedig megerősítik vagy finomítják ezeket az elméleti előrejelzéseket.
A kutatás egyik fő kihívása a kétféle eredmény közötti esetleges eltérések feloldása. Az elméleti modellek mindig tartalmaznak közelítéseket, és a kísérleti adatok is tartalmazhatnak mérési hibákat vagy torzításokat. Az összehasonlítás és a modell finomítása egy iteratív folyamat, amely hozzájárul a molekuláris szerkezet és dinamika egyre pontosabb képének kialakításához. Különösen a komplexebb rendszerek, mint például a polimerek, fehérjék vagy nukleinsavak konformációjának vizsgálata során merülnek fel jelentős kihívások, ahol a lehetséges konformációk száma exponenciálisan növekszik.
A félszék konformáció dinamikus természete, a gyors egyensúlyok, az átmeneti állapotok, valamint a környezeti tényezők és az elméleti modellek hatásának megértése a konformációs analízis élvonalbeli kutatásainak központjában áll, folyamatosan bővítve tudásunkat a molekulák térbeli viselkedéséről.
A jövőbeli kutatások várhatóan még pontosabb számítási módszereket, kifinomultabb spektroszkópiai technikákat és a mesterséges intelligencia (AI) alkalmazását fogják felhasználni a konformációs analízisben. Az AI és a gépi tanulás képes lehet nagy adathalmazok elemzésére, új konformerek felfedezésére és a konformációs átmenetek predikciójára, ezzel forradalmasítva a molekuláris szerkezet és funkció közötti összefüggések megértését.







