
A szerves kémia, különösen a sztereokémia, a molekulák térbeli elrendeződésének és annak tulajdonságokra gyakorolt hatásának tudománya. Ezen a területen belül kiemelten fontos szerepet játszanak a konformációk, amelyek az atomok egy adott molekulában elfoglalt különböző térbeli elrendeződéseit jelentik. Ezek az elrendeződések egymásba forgatással, kötéstörés nélkül alakíthatók át, szemben a konfigurációkkal, amelyek csak kémiai kötések felszakításával és újrakötésével változtathatók meg. A cikloalkánok, vagyis a telített gyűrűs szénhidrogének, különösen érdekes példát szolgáltatnak a konformációs analízisre, mivel a gyűrűs szerkezet speciális feszültségeket és korlátozásokat vezet be a molekula mozgásába. Ezen gyűrűs molekulák stabilitása, reakciókészsége és biológiai aktivitása nagymértékben függ attól, hogy milyen konformációkat vesznek fel a térben.
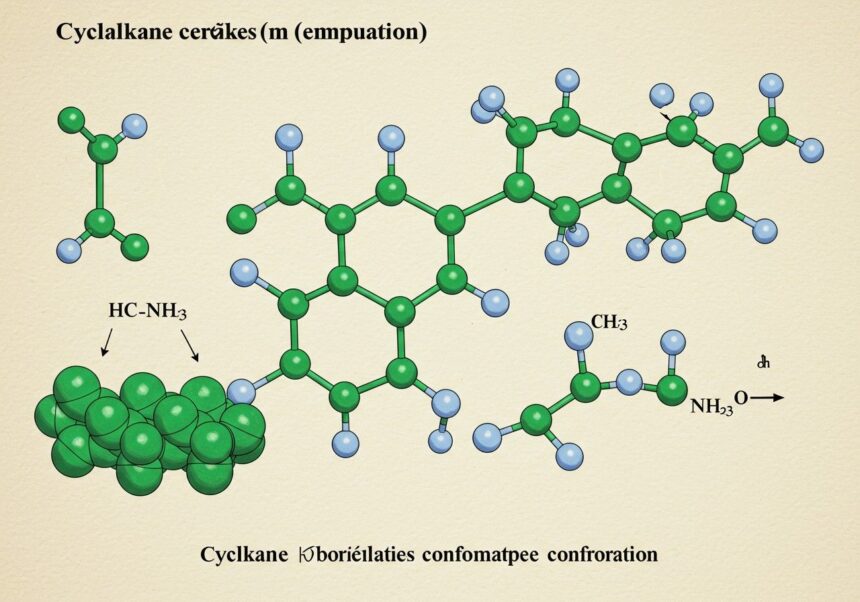
A boríték konformáció egy ilyen specifikus térbeli elrendeződés, amely különösen az ötös gyűrűs rendszerekre, mint például a ciklopentánra és számos biológiailag aktív molekula gyűrűjére jellemző. Ez a konformáció alapvetően befolyásolja az érintett molekulák kémiai és fizikai tulajdonságait, beleértve a reakciók szelektivitását és a gyógyszerek receptorokhoz való kötődését. A kémiai és biológiai rendszerek komplexitásának megértéséhez elengedhetetlen a molekulák dinamikus térbeli viselkedésének, így a boríték konformáció részletes ismerete.
A konformációs analízis célja, hogy megértse ezeket a térbeli elrendeződéseket, az azokat befolyásoló energiákat, és azt, hogy hogyan alakulnak át egymásba. A gyűrűs molekulák esetében a gyűrűfeszültség fogalma kulcsfontosságú. Ez a feszültség alapvetően két fő komponensből tevődik össze: a szögfeszültségből, amelyet Baeyer-feszültségként is ismerünk, és a torsziós feszültségből, amelyet Pitzer-feszültségnek is neveznek. A szögfeszültség abból ered, hogy az atomok közötti kötésszögek eltérnek az ideális értékektől (pl. a sp3 hibridizált szénatomok esetében a 109,5°-os tetraéderes szögtől). A torsziós feszültség pedig a szomszédos szénatomokon lévő szubsztituensek egymáshoz viszonyított elhelyezkedéséből fakad; az úgynevezett fedőállású (eclipsed) konformációk energiája magasabb, mint az eltolt (staggered) konformációké, az elektronfelhők taszítása miatt. A cikloalkánok ezért igyekeznek olyan konformációt felvenni, amely minimalizálja ezen feszültségeket, és így a lehető legalacsonyabb energiájú, legstabilabb állapotba kerülnek.
A boríték konformáció megértése elengedhetetlen a gyűrűs rendszerek, különösen a biológiailag fontos ötös gyűrűk, például a ribóz és dezoxiribóz viselkedésének értelmezéséhez. Ezek a molekulák alkotják a DNS és RNS gerincét, és konformációjuk kulcsfontosságú a nukleinsavak szerkezetében és funkciójában. A kémikusok és biokémikusok számára a boríték konformáció részletes ismerete alapvető fontosságú a molekuláris kölcsönhatások, a gyógyszertervezés és az anyagszerkezeti kutatások szempontjából, hiszen ez a térbeli elrendeződés jelentősen befolyásolja a molekulák reakciókészségét és biológiai célpontokkal való interakcióját.
A cikloalkánok konformációjának alapjai és a gyűrűfeszültség
A cikloalkánok, mint a ciklopentán, ciklohexán és más gyűrűs szénhidrogének, térbeli szerkezete messze nem triviális. Míg a nyílt láncú alkánok viszonylag szabadon foroghatnak a szén-szén kötések mentén, a gyűrűs szerkezet jelentősen korlátozza ezt a mozgást. Ennek ellenére a gyűrűs molekulák sem merevek; dinamikusak, és különböző konformációk között képesek átalakulni. Ezen átalakulások során a molekula energiája változik, és a legalacsonyabb energiájú állapotok a legstabilabb konformációk.
A gyűrűfeszültség fogalmát Adolf von Baeyer vezette be a 19. század végén. Baeyer feltételezte, hogy a cikloalkánok sík szerkezetűek, és a gyűrűfeszültség abból adódik, hogy a kötésszögek eltérnek az ideális 109,5°-os tetraéderes szögtől. Elmélete szerint a ciklopropán (60°), ciklobután (90°) és ciklohexán (120°) jelentős szögfeszültséggel rendelkezne, míg a ciklopentán (108°) minimális eltérést mutatna. Azonban a valóságban a ciklohexán a legstabilabb cikloalkán, ami azt jelezte, hogy Baeyer elmélete nem volt teljes, mivel nem vette figyelembe a torsziós feszültséget.
Később kiderült, hogy a cikloalkánok nem sík szerkezetűek, hanem úgynevezett puckeringet, azaz gyűrűtorzulást mutatnak. Ez a torzulás lehetővé teszi, hogy a molekula enyhítse a Baeyer által felismert szögfeszültséget és a később felismert torsziós feszültséget. A torsziós feszültség a szomszédos szénatomokon lévő hidrogénatomok (vagy más szubsztituensek) közötti fedőállású kölcsönhatásokból ered. Ideális esetben az eltolt állás minimalizálja ezt a feszültséget, mivel az elektronfelhők taszítása a legkisebb. Például az etán esetében az eltolt konformáció körülbelül 3 kcal/mol-lal stabilabb, mint a fedőállású.
A különböző méretű cikloalkánok eltérő módon oldják meg ezt a feszültségminimalizálási problémát. A ciklobután például egy „pillangó” konformációt vesz fel, ahol a gyűrű két szénatomja kiemelkedik a másik kettő síkjából. A ciklohexán a jól ismert szék konformációt preferálja, amelyben a szögfeszültség és a torsziós feszültség is minimális, mivel minden kötésszög közel ideális 109,5°-os, és minden szomszédos hidrogén eltolt állásban van. Ez a szék konformáció rendkívül stabil, és a ciklohexán a szerves kémia egyik legfontosabb modellmolekulája a konformációs analízis tanulmányozásában.
„A cikloalkánok térbeli szerkezetének megértése kulcsfontosságú a molekulák stabilitásának, reakciókészségének és biológiai aktivitásának előrejelzéséhez, hiszen a konformáció alapvetően befolyásolja a kémiai tulajdonságokat.”
A ciklopentán esete azonban különleges, és itt lép be a képbe a boríték konformáció. Mivel öt szénatomja van, a szék konformáció nem lehetséges a gyűrű bezárása miatt. Egy sík ciklopentán gyűrűben a kötésszögek 108°-osak lennének, ami nagyon közel van az ideális 109,5°-hoz, így a szögfeszültség minimális lenne. Azonban egy sík ciklopentánban az összes szomszédos hidrogénatom fedőállásban lenne, ami jelentős torsziós feszültséget eredményezne (körülbelül 10 kcal/mol, ami a ciklohexán csónak konformációjához hasonló érték). Ennek a torsziós feszültségnek az enyhítése érdekében a ciklopentán torzult, nem sík formát vesz fel.
A boríték konformáció részletes vizsgálata
A boríték konformáció a ciklopentán és hasonló ötös gyűrűs rendszerek jellegzetes térbeli elrendeződése. Nevét onnan kapta, hogy a molekula egy kinyitott borítékra emlékeztet, ahol a gyűrű öt atomjából négy nagyjából egy síkban helyezkedik el, míg az ötödik atom kiemelkedik ebből a síkból, mint egy lepecsételt boríték füle. Ez a „kiemelkedő” atom eltér a többi négy síkjától, és éppen ez a kiemelkedés teszi lehetővé a torsziós feszültség minimalizálását a szögfeszültség minimális növekedése árán.
A ciklopentán és a boríték konformáció
A ciklopentán gyűrűje öt szénatomból áll. Egy hipotetikus sík ciklopentán molekulában minden C-C-C kötésszög 108° lenne, ami nagyon közel van az ideális 109,5°-os tetraéderes szöghöz, tehát a szögfeszültség minimális lenne. Azonban ebben a sík elrendeződésben az összes szomszédos C-H kötés fedőállásban lenne, ami jelentős torsziós feszültséget okozna (kb. 10 kcal/mol, hasonlóan a ciklohexán csónak konformációjához). Ennek a torsziós feszültségnek az enyhítése érdekében a ciklopentán nem sík konformációt vesz fel.
A ciklopentán két fő nem sík konformációt vehet fel: a boríték (envelope, E) konformációt és a fél-szék (half-chair, T) konformációt (más néven twist vagy csavart konformáció). Mindkét konformáció minimalizálja a torsziós feszültséget a sík formához képest, de a szögfeszültség növekedésével jár. A boríték konformációban egy szénatom (a „flap” atom) kiemelkedik a többi négy szénatom által alkotott síkból. A fél-szék konformációban két szomszédos szénatom emelkedik ki a többiek síkjából, de ellenkező irányba, ami egy csavart formát eredményez.
A ciklopentán esetében a boríték és a fél-szék konformációk energiája nagyon hasonló, és a molekula dinamikusan, gyorsan átalakul ezek között. Ez a jelenség az úgynevezett pszeudorotáció. A pszeudorotáció során a „kiemelkedő” atom a gyűrűben körbejár, mintha forogna, de valójában az atomok helyzete változik a gyűrűben, és folyamatosan új boríték vagy fél-szék konformációk jönnek létre. Ez azt jelenti, hogy a ciklopentán nem egyetlen stabil konformációban létezik, hanem egy „konformációs sokaság” része, ahol az energiagátak az átalakulások között nagyon alacsonyak (körülbelül 1-2 kcal/mol, ami szobahőmérsékleten nagyon gyors átmenetet tesz lehetővé).
A pszeudorotáció következtében a ciklopentán molekula folyamatosan változtatja alakját, ami szobahőmérsékleten nagyon gyors folyamat. Ezért a hagyományos NMR spektroszkópia nem képes megkülönböztetni a különböző hidrogénatomokat, mivel azok átlagolt környezetet látnak. Alacsony hőmérsékleten (pl. -100 °C alatt) azonban, amikor a pszeudorotáció lelassul, már megfigyelhetők a különbségek, és a hidrogénatomok jelei szétválnak a spektrumban.
A boríték konformáció jellemzői
A boríték konformációt a következőképpen jellemezhetjük:
- Egyetlen kiemelkedő atom: A gyűrű öt atomjából egy (az ún. „flap” atom) emelkedik ki a másik négy atom által definiált síkból. Ez a sík általában a gyűrű síkját alkotó atomok között húzódik.
- Minimalizált torsziós feszültség: A kiemelkedés lehetővé teszi a szomszédos szénatomokon lévő szubsztituensek közötti fedőállású kölcsönhatások enyhítését, gyakran eltolt vagy közel eltolt állásba kényszerítve őket. Ez az elsődleges hajtóerő a boríték konformáció kialakulásában.
- Növelt szögfeszültség: A boríték konformációban a kötésszögek kissé torzulnak az ideális 109,5°-tól, de ez a szögfeszültség növekedés kisebb, mint a torsziós feszültség csökkenése, így az összenergia alacsonyabb, mint a sík formában. Az optimális geometria a két feszültségtípus közötti kompromisszum.
- Dinamikus természet (pszeudorotáció): A boríték konformációk gyorsan átalakulnak egymásba a pszeudorotáció mechanizmusa révén, ahol az „flap” atom vándorol a gyűrűben. Ez a jelenség a ciklopentánra és számos ötös gyűrűs heterociklusra jellemző.
A boríték konformáció energiája a sík konformációhoz képest körülbelül 5-6 kcal/mol-lal alacsonyabb, ami jelentős stabilitásnövekedést jelent. A fél-szék konformáció energiája nagyon hasonló, néha minimálisan alacsonyabb vagy magasabb, attól függően, hogy milyen szubsztituensek vannak a gyűrűn, vagy hogy milyen heterociklusos atomok épülnek be. A szubsztituensek bevezetése a gyűrűbe gyakran „preferált” boríték vagy fél-szék konformációt eredményezhet, mivel a nagyobb csoportok sterikus kölcsönhatásai egy adott térbeli elrendezést stabilizálnak.
Boríték konformáció más cikloalkánokban és heterociklusokban
Bár a boríték konformáció legjellegzetesebben a ciklopentánnal és származékaival hozható összefüggésbe, a puckering jelensége és a torzult gyűrűs formák általánosak a gyűrűs rendszerekben. Fontos megjegyezni, hogy a „boríték” elnevezést szigorúan az ötös gyűrűkre használják, ahol egyetlen atom emelkedik ki a síkból. Más gyűrűméretek esetén eltérő, de hasonló elven alapuló torzult konformációk figyelhetők meg, amelyek szintén a feszültségek minimalizálását szolgálják.
Ciklobután: a pillangó konformáció
A ciklobután egy négytagú gyűrű. Egy sík ciklobután gyűrűben a belső kötésszögek 90°-osak lennének, ami jelentős szögfeszültséget okozna (az ideális 109,5°-hoz képest 19,5°-os eltérés atomonként). Emellett az összes szomszédos hidrogén fedőállásban lenne, ami nagy torsziós feszültséget eredményezne. A ciklobután ezért nem sík formát vesz fel, az úgynevezett pillangó (butterfly) konformációt. Ebben a konformációban a gyűrű két átellenes szénatomja kiemelkedik a másik két szénatom által alkotott síkból, ellenkező irányba. Ez a torzulás enyhíti a torsziós feszültséget, de növeli a szögfeszültséget. A ciklobután esetében a szögfeszültség dominánsabb, mint a torsziós feszültség, de a pillangó konformáció az optimális kompromisszum, körülbelül 6 kcal/mol-lal stabilabb a sík formánál. A pillangó konformációk között is van dinamikus átmenet, bár az energiagát magasabb, mint a ciklopentán esetében.
Ciklohexán: a szék, csónak és fél-szék
A ciklohexán a szerves kémia „királya” a konformációs analízis szempontjából, mivel a szék konformációja gyakorlatilag feszültségmentes. Ebben a konformációban minden kötésszög közel ideális 109,5°, és minden szomszédos hidrogén eltolt állásban van. A szék konformáció azonban dinamikusan átalakulhat egy másik szék konformációvá az úgynevezett gyűrű inverzió (ring flip) révén. Ez a folyamat egy energiagáton keresztül megy végbe, amelynek átmeneti állapota a fél-szék konformáció. A ciklohexán esetében a fél-szék konformációban négy szénatomot tartalmaz egy síkban, és a fennmaradó két szénatom kiemelkedik ebből a síkból, egy szék és egy csónak konformáció közötti átmeneti formát képezve. A csónak konformáció is létezik, de magasabb energiájú a szék konformációhoz képest a fedőállású hidrogének és a „zászlóállású” hidrogének közötti sterikus taszítás miatt. Bár nem boríték, a fél-szék is egy példa a gyűrűs rendszerek torzult, nem sík formáira, melyek a feszültség minimalizálását szolgálják, vagy átmeneti állapotként jelennek meg.
Nagyobb cikloalkánok: komplexebb konformációk
A nagyobb cikloalkánok, mint a cikloheptán (7 tagú), ciklooktán (8 tagú), vagy ciklononán (9 tagú), még komplexebb konformációs viselkedést mutatnak. Ezek a gyűrűk számos lehetséges konformációt vehetnek fel, amelyek energiája nagyon hasonló. A gyűrűméret növekedésével a transzannuláris feszültségek (a gyűrűn belüli, nem szomszédos atomok közötti kölcsönhatások, pl. sterikus taszítások) is jelentőssé válnak. Ezek a kölcsönhatások tovább bonyolítják a stabil konformációk előrejelzését.
A cikloheptán például felvehet csavart-szék (twist-chair), csavart-csónak (twist-boat) és szék (chair) konformációkat, melyek között gyors átmenetek zajlanak. A ciklooktán még ennél is sokfélébb, többek között korona (crown), csavart-szék (twist-chair), és csavart-csónak (twist-boat) konformációkat is mutathat. Ezen konformációk energiakülönbségei rendkívül kicsik, gyakran csak néhány tized kcal/mol, ami azt jelenti, hogy szobahőmérsékleten folyamatosan átalakulnak egymásba, és egy átlagolt szerkezetet figyelhetünk meg.
Ezekben a nagyobb gyűrűkben a „boríték” elnevezést ritkán használják, de az alapelv, miszerint a gyűrű torzul a feszültségek minimalizálása érdekében, továbbra is érvényes. A torzulás mértéke és jellege azonban sokkal változatosabb, és a stabil konformációk az egyensúlyi geometriák széles skáláját ölelik fel. A pontos konformáció meghatározása ezekben az esetekben gyakran csak kifinomult számítógépes modellezéssel és alacsony hőmérsékletű spektroszkópiai technikákkal lehetséges.
Heterociklusos rendszerek: a biológiai jelentőség
A boríték konformáció különösen nagy jelentőséggel bír a heterociklusos rendszerekben, ahol a gyűrűben a szénatomok mellett más atomok (pl. oxigén, nitrogén, kén) is előfordulnak. Az ötös gyűrűs heterociklusok, mint a furanóz (oxigént tartalmazó ötös gyűrű) és pirrolidin (nitrogént tartalmazó ötös gyűrű) gyűrűk, gyakran boríték vagy fél-szék konformációt vesznek fel. Ezek a gyűrűk alapvető építőkövei számos biológiailag fontos molekulának, és konformációjuk kulcsfontosságú a biológiai funkciók szempontjából.
A legkiemelkedőbb példák a ribóz és a dezoxiribóz, amelyek a nukleinsavak (RNS és DNS) cukor komponensei. A ribóz és dezoxiribóz ötös gyűrűje furanóz formában létezik (az oxigénatom is része a gyűrűnek), és ezek a gyűrűk gyakran boríték vagy fél-szék konformációt vesznek fel. A konformációjuk kulcsfontosságú a nukleotidok és nukleinsavak térbeli szerkezetében, valamint a DNS és RNS kettős hélixének stabilitásában és működésében. A cukorgyűrű puckeringje, azaz torzulása, alapvetően befolyásolja a foszfodiészter gerinc térbeli elrendeződését és a bázisok egymáshoz viszonyított pozícióját.
Például, a DNS-ben a dezoxiribóz gyűrűje általában C2′-endo (vagy 2E boríték) vagy C3′-endo (vagy 3E boríték) konformációban található, ahol a C2′ vagy a C3′ szénatom emelkedik ki a gyűrű síkjából (endo jelentése, hogy a kiemelkedés a bázis felé történik). Ezek a finom konformációs különbségek befolyásolják a foszfodiészter gerinc térbeli elrendeződését, és hozzájárulnak a DNS különböző formáinak (A-DNS, B-DNS, Z-DNS) kialakulásához. A B-DNS, a legelterjedtebb forma, jellemzően C2′-endo puckeringet mutat, míg az A-DNS és az RNS gyakrabban C3′-endo ribóz gyűrűkkel rendelkezik. Ezek a különbségek határozzák meg a hélix geometriáját, a bázisok közötti távolságot és a hélix hornyainak mélységét, amelyek mind kritikusak a fehérjékkel való kölcsönhatásban és a genetikai információ átadásában.
„A biológiailag aktív molekulák, mint a cukrok és nukleozidok konformációja alapvetően meghatározza azok funkcióját és kölcsönhatásait az élő rendszerekben, a gyógyszerhatástól a genetikai információ átadásáig.”
Egy másik fontos példa a prolin aminosav, amelynek pirrolidin gyűrűje (egy nitrogént tartalmazó ötös gyűrű) szintén boríték konformációt vesz fel. A prolin gyűrűjének merevsége és specifikus konformációja jelentősen befolyásolja a fehérjék másodlagos és harmadlagos szerkezetét, például a kollagén hélixében, ahol a prolin gyakran fordul elő. A prolin gyűrűje korlátozza a peptidkötés körüli forgást, ami „törést” okozhat a fehérje szerkezetében, és kulcsfontosságú a fehérjék hajtogatásában és térbeli szerkezetének kialakításában. Számos más biológiailag aktív molekula, például szteroidok, alkaloidok és antibiotikumok is tartalmaznak ötös gyűrűket, amelyek boríték konformációban létezhetnek, és amelyek szerkezeti rugalmassága és specificitása alapvető fontosságú biológiai hatásukhoz.
A konformáció stabilitását befolyásoló tényezők

A boríték konformáció, ahogy más cikloalkán konformációk is, számos tényező komplex kölcsönhatásának eredményeként alakul ki. Ezek a tényezők határozzák meg a molekula preferált térbeli elrendeződését és dinamikáját, valamint az egyes konformerek relatív energiáját.
1. Szögfeszültség (Baeyer-feszültség)
Ez a feszültség akkor keletkezik, ha a kötésszögek eltérnek az ideális értékektől (pl. 109,5° a sp3 hibridizált szénatomok esetében). Egy sík cikloalkánban a kötésszögek matematikailag meghatározottak (pl. 60° a ciklopropánban, 90° a ciklobutánban, 108° a ciklopentánban, 120° a ciklohexánban). Minél nagyobb az eltérés az ideálistól, annál nagyobb a szögfeszültség. A boríték konformációban a gyűrű torzulása enyhíti a torsziós feszültséget, de gyakran kissé növeli a szögfeszültséget a sík formához képest. Az optimális konformáció az, amelyik a két feszültségtípus közötti legjobb kompromisszumot jelenti, a molekula minimális összenergiával rendelkezik.
2. Torsziós feszültség (Pitzer-feszültség)
Ez a feszültség a szomszédos atomokon lévő szubsztituensek közötti fedőállású (eclipsed) kölcsönhatásokból ered. Az eltolt (staggered) állású konformációk energiája alacsonyabb, mivel a szubsztituensek távolabb vannak egymástól, minimalizálva az elektronfelhők taszítását. Egy sík ciklopentánban az összes C-H kötés fedőállásban lenne, ami hatalmas torsziós feszültséget jelentene. A boríték konformációban az atomok kiemelkedése a síkból lehetővé teszi, hogy a szomszédos C-H kötések eltoltabb állásba kerüljenek, csökkentve ezzel a torsziós feszültséget. Ez a torsziós feszültség a ciklopentán esetében a domináns tényező, amely a nem sík konformációk felvételét kényszeríti.
3. Sterikus feszültség (van der Waals taszítás)
Ez a feszültség akkor jelentkezik, ha két atom vagy atomcsoport túl közel kerül egymáshoz, és elektronfelhőik taszítani kezdik egymást. Ez különösen szubsztituált cikloalkánok esetében fontos. Például a metilcsoportok közötti 1,3-diaxiális kölcsönhatás a ciklohexánban jelentős sterikus feszültséget okozhat. A boríték konformációban a szubsztituensek elhelyezkedése is befolyásolja az energiát. Egy nagy szubsztituens általában az „equatorial” (ekvatoriális) jellegű pozíciót preferálja, azaz a gyűrű síkjához közelebb eső, kifelé mutató pozíciót, hogy minimalizálja a sterikus gátlást a gyűrűn belüli hidrogénatomokkal vagy más szubsztituensekkel.
4. Transzannuláris feszültség (nagyobb gyűrűkben)
A nagyobb gyűrűkben (7 tagú vagy annál nagyobb) a gyűrűn belüli, nem szomszédos atomok vagy szubsztituensek közötti kölcsönhatások is jelentőssé válnak. Ezek a transzannuláris kölcsönhatások lehetnek taszítóak (sterikus gátlás) vagy vonzóak (pl. hidrogénkötések), és befolyásolják a preferált konformációt. A transzannuláris feszültségek miatt a nagyobb gyűrűk gyakran komplexebb, sokféle konformációt vesznek fel, melyek energiája nagyon közel áll egymáshoz.
5. Elektronikus hatások és anomer effektus
Heterociklusos rendszerekben, különösen az oxigént vagy nitrogént tartalmazó gyűrűkben (pl. cukrok), elektronikus hatások, mint az anomer effektus, szintén befolyásolhatják a konformáció stabilitását. Az anomer effektus a heteroatomokhoz közeli szubsztituensek preferált axiális vagy ekvatoriális elhelyezkedésére utal, amely a kötéspályák közötti kölcsönhatásokból ered. Ez a hatás stabilizálhat bizonyos boríték vagy fél-szék konformációkat a cukorgyűrűkben.
6. Dipól-dipól kölcsönhatások és hidrogénkötések
Ha a gyűrűn poláris szubsztituensek találhatók, a dipól-dipól kölcsönhatások és a belső hidrogénkötések is szerepet játszhatnak a konformáció stabilizálásában. Ezek a kölcsönhatások specifikus térbeli elrendeződéseket favorizálhatnak, például egy intramolekuláris hidrogénkötés stabilizálhat egy olyan konformációt, amelyben a donor és akceptor csoportok közel kerülnek egymáshoz. A külső környezet, például a oldószer polaritása is befolyásolhatja ezeket a kölcsönhatásokat és ezáltal a konformációs egyensúlyt.
A konformációs analízis módszerei és a boríték konformáció detektálása
A konformációk, köztük a boríték konformáció, tanulmányozása számos kísérleti és elméleti módszerrel történik. Ezek a módszerek lehetővé teszik a molekulák térbeli szerkezetének meghatározását, az energiagátak mérését az átalakulások között, és a dinamikus viselkedés megértését, mind oldatban, mind szilárd fázisban vagy gázfázisban.
1. Nukleáris Mágneses Rezonancia (NMR) Spektroszkópia
Az NMR spektroszkópia az egyik legerősebb eszköz a molekulák szerkezetének és dinamikájának vizsgálatára oldatban. Mivel a különböző konformációkban a hidrogénatomok (vagy más NMR-aktív magok, pl. 13C, 19F) eltérő kémiai környezetben helyezkednek el, az NMR spektrumok információt szolgáltathatnak róluk. A kémiai eltolódások és a csatolási állandók (különösen a vicinális protonok közötti 3JHH csatolások, melyeket a Karplus-egyenlet ír le, és a diéderes szögtől függenek) alapvető betekintést nyújtanak a kötések körüli térbeli elrendeződésbe.
Szobahőmérsékleten, ha a konformációs átalakulások gyorsak (mint a ciklopentán pszeudorotációja), a spektrum átlagolt jeleket mutat, ami azt jelzi, hogy a molekula gyorsan fluktuál a különböző konformerek között. Azonban alacsonyabb hőmérsékleteken, amikor az átalakulások lelassulnak, az egyes konformációk jelei szétválnak, lehetővé téve azok azonosítását és az energiagátak meghatározását az átalakulásokhoz. A 2D NMR technikák, mint a NOESY (Nuclear Overhauser Effect Spectroscopy) és COSY (Correlation Spectroscopy) kísérletekkel a hidrogénatomok térbeli közelsége is megállapítható, ami a konformáció felderítéséhez elengedhetetlen, különösen komplex molekulák esetében.
2. Röntgendiffrakció
A röntgendiffrakció (XRD) kristályos állapotban lévő molekulák pontos 3D szerkezetét képes meghatározni, atomi felbontásban. Ez a módszer „pillanatfelvételt” készít a molekuláról annak kristályrácsban elfoglalt pozíciójában. Ha egy ciklopentán-származék kristályos formában létezik, a röntgendiffrakció egyértelműen kimutathatja a boríték konformációt, megadva a kiemelkedő atom pozícióját, a kötésszögeket és a torsziós szögeket. Azonban fontos megjegyezni, hogy a kristályos állapotban megfigyelt konformáció nem feltétlenül azonos az oldatban vagy gázfázisban preferált konformációval, mivel a kristályrács erői (ún. kristálypakolási effektusok) befolyásolhatják a molekula alakját és stabilizálhatnak egy egyébként kevésbé preferált konformációt.
3. Elektron diffrakció és mikrohullámú spektroszkópia
Gázfázisú molekulák esetében az elektron diffrakció és a mikrohullámú spektroszkópia is használható a konformációk meghatározására. Ezek a módszerek különösen hasznosak kis, illékony molekulák, például egyszerű cikloalkánok esetén, ahol a molekulák szabadon, külső kölcsönhatásoktól mentesen viselkednek. Az elektron diffrakció a molekulák átlagos geometriájáról nyújt információt, míg a mikrohullámú spektroszkópia a molekulák rotációs átmeneteit vizsgálja, amelyek érzékenyek a molekula tehetetlenségi nyomatékára, így a pontos térbeli szerkezetére.
4. Számítógépes kémia és molekuladinamika
A számítógépes kémiai módszerek, mint a molekulamechanika (MM), a sűrűségfunkcionál-elmélet (DFT), és az ab initio számítások, kulcsfontosságúak a konformációs analízisben. Ezek a módszerek képesek előre jelezni a molekulák stabil konformációit (az energia minimumokat), az energiagátakat az átalakulások között (az átmeneti állapotok energiáját), és a molekulák dinamikus viselkedését. A molekuladinamikai szimulációk lehetővé teszik a molekulák mozgásának és konformációs változásainak időbeli követését, feltárva a pszeudorotáció mechanizmusát és a különböző konformerek közötti gyors átmeneteket. Ezek az elméleti eszközök kiegészítik a kísérleti adatokat, és mélyebb betekintést nyújtanak a molekulák térbeli viselkedésébe, különösen olyan esetekben, ahol a kísérleti adatok nehezen értelmezhetők vagy hiányosak.
Az ötös gyűrűk, mint a ciklopentán vagy a furanóz, konformációs állapotának leírására gyakran használnak úgynevezett puckering koordinátákat (pl. Cremer-Pople paraméterek). Ezek a matematikai paraméterek lehetővé teszik a gyűrű torzulásának kvantitatív jellemzését, és megkülönböztetik a különböző boríték és fél-szék formákat, valamint a pszeudorotációs fázist. Ez a kvantitatív leírás elengedhetetlen a konformációs átalakulások mechanizmusának és az azokat befolyásoló tényezőknek a részletes megértéséhez.
A boríték konformáció jelentősége a reakciókészségben és a biokémiában
A molekulák térbeli elrendeződése, így a boríték konformáció is, messzemenő hatással van azok kémiai reakciókészségére, szelektivitására és biológiai aktivitására. A sztereokémia nem csupán elméleti érdekesség, hanem a gyakorlati kémia, a gyógyszerfejlesztés és a biológiai folyamatok alapköve.
1. Reakciókészség és szelektivitás
A gyűrűs rendszerek konformációja befolyásolja a szubsztituensek térbeli hozzáférhetőségét, ami kritikus lehet a reakciók kimenetele szempontjából. Például egy adott nukleofil támadás vagy eliminációs reakció csak akkor mehet végbe hatékonyan, ha a reaktánsok a megfelelő térbeli orientációban tudnak találkozni. A boríték konformációban lévő ötös gyűrűk merevebbek, mint a nyílt láncú analógjaik, de rugalmasabbak, mint a ciklohexán szék konformációja. Ez a rugalmasság lehetővé teszi, hogy a molekula könnyebben alkalmazkodjon a reakcióátmeneti állapotokhoz, de a specifikus boríték forma mégis irányíthatja a sztereoszelektivitást.
Például, ha egy szubsztituált ciklopentán-származék boríték konformációt vesz fel, a kiemelkedő atomhoz kapcsolódó szubsztituensek eltérő térbeli helyzetet fognak elfoglalni, mint a síkban lévők. Ez befolyásolhatja a reagens „hozzáférését” a gyűrűhöz, és így a reakció sztereokémiai kimenetelét. Ez különösen fontos lehet a szintézisekben, ahol specifikus sztereoizomerek előállítása a cél. A reakciók sebességét és termékszelekcióját is befolyásolja a molekula konformációs egyensúlya, mivel a reakció gyakran egy adott konformeren keresztül megy végbe.
2. Gyógyszertervezés és receptor kölcsönhatások
A gyógyszermolekulák gyakran tartalmaznak gyűrűs szerkezeteket, köztük ötös gyűrűket. A gyógyszerek hatásmechanizmusa gyakran azon alapul, hogy képesek specifikus receptorokhoz vagy enzimekhez kötődni. Ez a kötődés rendkívül sztereospecifikus, ami azt jelenti, hogy a gyógyszermolekula térbeli alakja pontosan illeszkedik a receptor kötőhelyéhez, mint egy kulcs a zárba. A boríték konformációban lévő gyűrűk térbeli elrendeződése alapvetően meghatározza a molekula „alakját” és így a receptorhoz való affinitását és szelektivitását. A konformációs rugalmasság lehetővé teszi a gyógyszerek számára, hogy adaptálódjanak a receptor kötőhelyéhez (ún. indukált illeszkedés modellje), de a stabil konformerek ismerete alapvető a tervezéshez.
A gyógyszertervezés során a kutatók gyakran használnak számítógépes modellezést (docking szimulációkat) annak előrejelzésére, hogy egy molekula hogyan illeszkedik egy receptorhoz. Ezek a szimulációk figyelembe veszik a molekula konformációs rugalmasságát, beleértve a boríték konformáció felvételének lehetőségét is. A stabil boríték konformációk ismerete segíthet optimalizálni a gyógyszermolekulák szerkezetét a jobb hatékonyság és kevesebb mellékhatás érdekében. Például, a nukleozid analóg gyógyszerek, amelyeket vírusellenes és rákellenes terápiákban használnak, gyakran módosított ribóz vagy dezoxiribóz gyűrűkkel rendelkeznek, amelyek konformációja kritikus a gyógyszer célpont enzimhez való kötődéséhez és aktivitásához.
3. Biokémiai folyamatok és biológiai makromolekulák
Ahogy korábban említettük, a ribóz és dezoxiribóz, a nukleinsavak cukor komponensei, gyakran boríték konformációt vesznek fel. A C2′-endo és C3′-endo boríték formák közötti különbség alapvetően befolyásolja a DNS és RNS kettős hélixének szerkezetét és stabilitását. A B-DNS, a legelterjedtebb DNS forma, tipikusan C2′-endo dezoxiribózt tartalmaz, míg az A-DNS és az RNS gyakran C3′-endo ribózt. Ezek a finom konformációs különbségek határozzák meg a hélix geometriáját, a bázisok közötti távolságot és a hélix hornyainak mélységét, amelyek mind kritikusak a fehérjékkel való kölcsönhatásban, a DNS replikációjában, transzkripciójában és a genetikai információ átadásában.
A prolin aminosav, mint egy gyűrűs aminosav, szintén boríték konformációt vesz fel a pirrolidin gyűrűjében. Ez a konformáció korlátozza a prolin peptidkötés körüli forgását, ami egyedi szerkezeti tulajdonságokat kölcsönöz a fehérjéknek, különösen a kollagénnek, amelyben a prolin és hidroxiprolin magas aránya miatt stabil hélixek alakulnak ki. A prolin jelenléte a fehérjeláncban „törést” okozhat a hélix szerkezetben, és kulcsfontosságú a fehérjék hajtogatásában és térbeli szerkezetének kialakításában, befolyásolva az enzimek aktivitását és a fehérje-fehérje kölcsönhatásokat.
Sok más biológiailag aktív molekula, például szteroidok (melyek gyakran tartalmaznak fúziós ötös gyűrűket), alkaloidok és antibiotikumok is tartalmaznak ötös gyűrűket, amelyek boríték konformációban létezhetnek. Ezen molekulák térbeli elrendeződésének pontos ismerete elengedhetetlen a biológiai mechanizmusok megértéséhez és új terápiás szerek fejlesztéséhez, valamint a biológiai rendszerek dinamikus molekuláris kölcsönhatásainak feltárásához.
Összefoglaló táblázat: Cikloalkán konformációk és feszültségek
Az alábbi táblázat összefoglalja a legfontosabb cikloalkánok preferált konformációit és az azokat befolyásoló feszültségtípusokat, kiemelve a boríték konformáció helyét a gyűrűs rendszerek konformációs spektrumában.
| Cikloalkán | Gyűrűméret | Preferált konformáció | Domináns feszültség(ek) | Megjegyzés |
|---|---|---|---|---|
| Ciklopropán | 3 | Sík | Szögfeszültség, Torsziós feszültség | Rendkívül nagy szögfeszültség (60° kötésszög). Kémiailag nagyon reaktív. |
| Ciklobután | 4 | Pillangó (butterfly) | Szögfeszültség, Torsziós feszültség | A sík forma szögfeszültsége és torsziós feszültsége is magas. A pillangó enyhíti a torsziós feszültséget. |
| Ciklopentán | 5 | Boríték (envelope), Fél-szék (half-chair) | Torsziós feszültség, Szögfeszültség | Sík forma torsziós feszültsége magas. A boríték és fél-szék enyhíti ezt, pszeudorotáció jellemzi. |
| Ciklohexán | 6 | Szék (chair) | Minimális | Feszültségmentes gyűrű, ideális kötésszögek és eltolt állású hidrogének. Gyűrű inverzió. |
| Cikloheptán | 7 | Csavart-szék (twist-chair) | Torsziós feszültség, Transzannuláris feszültség | Több alacsony energiájú konformáció, komplex dinamika és gyors átmenetek. |
| Ciklooktán | 8 | Korona (crown), Csavart-szék (twist-chair), Csavart-csónak (twist-boat) | Torsziós feszültség, Transzannuláris feszültség | Még több lehetséges konformáció, transzannuláris kölcsönhatások jelentősek, rendkívül dinamikus. |
Konformációs dinamika és energia profilok

A konformációk közötti átalakulások nem azonnaliak, hanem egy energiagáton keresztül mennek végbe. Ennek az energiagátnak a magassága határozza meg az átalakulás sebességét, az Arrhenius-egyenlet szerint. A boríték konformáció esetében a ciklopentán pszeudorotációja egy nagyon alacsony energiagáton keresztül történik (kb. 1-2 kcal/mol), ami rendkívül gyors átmenetet eredményez a különböző boríték és fél-szék formák között. Ez a dinamikus viselkedés alapvetően befolyásolja a molekula átlagos térbeli elrendeződését és az oldatban megfigyelhető tulajdonságait.
Egy tipikus konformációs energia profil diagramon a stabil konformációk lokális energiagátakban helyezkednek el (minimumok), míg az átmeneti állapotok a maximumokon. A ciklopentán esetében a pszeudorotációs útvonal egy viszonylag lapos energiafelületen mozog, ahol a boríték és fél-szék formák energia minimumai nagyon közel vannak egymáshoz, és az átmeneti állapotok energiája is csak minimálisan magasabb. Ez magyarázza a gyors átmeneteket és a szobahőmérsékleten „átlagolt” szerkezetet, amelyet például az NMR spektroszkópia is detektál.
A szubsztituensek bevezetése a ciklopentán gyűrűbe megváltoztathatja ezt az energia profilt. Egy nagy szubsztituens stabilizálhat egy adott boríték vagy fél-szék konformációt azáltal, hogy a szubsztituens a legkevésbé sterikusan gátolt pozícióba kerül. Ezáltal a pszeudorotáció lelassulhat, vagy egy preferált konformáció dominánssá válhat, ami lehetővé teszi a specifikus konformációk detektálását és tanulmányozását NMR-rel vagy röntgendiffrakcióval. Például, a 1,2-dimetilciklopentán esetében a cisz- és transz-izomerek eltérő konformációs preferenciákat mutatnak a metilcsoportok közötti sterikus kölcsönhatások miatt, melyek egyedi boríték formákat stabilizálnak.
A konformációs dinamika nem csupán a stabilitást érinti, hanem a reakciókinetikát is. Egy molekula reakciókészsége gyakran függ attól, hogy milyen könnyen tudja felvenni a reakcióátmeneti állapotnak megfelelő konformációt. Ha az átmeneti állapot energiája magas egy adott konformációból kiindulva, akkor a reakció lassúbb lesz. Ezért a konformációs analízis alapvető fontosságú a reakciómechanizmusok megértésében és a kémiai szintézisek tervezésében, különösen a sztereoszelektív szintézisek esetében, ahol a termék térbeli elrendeződése kritikus.
A cikloalkánok, és különösen a ciklopentán boríték konformációja, a szerves kémia egyik legérdekesebb és legfontosabb területe. A gyűrűfeszültségek, a torsziós és szögfeszültségek közötti kényes egyensúly, valamint a molekulák dinamikus viselkedése mély betekintést enged a molekuláris szintű interakciókba. A boríték konformáció nem csupán egy kémiai érdekesség, hanem alapvető szerkezeti elem számos biológiailag aktív molekulában, melyek funkcióját és kölcsönhatásait alapvetően meghatározza. A modern spektroszkópiai és számítógépes kémiai módszerek révén egyre pontosabban feltárható ezen konformációk természete és szerepe, hozzájárulva a gyógyszerfejlesztéshez, anyagtudományhoz és az életfolyamatok alapvető megértéséhez, ami a jövőbeni kutatások egyik kulcsterülete marad.







