
A kémia világában a komplex vegyületek, vagy más néven koordinációs vegyületek, rendkívül sokszínű és jelentős csoportot alkotnak. Ezek a vegyületek központi fémionból és körülötte elhelyezkedő ligandumokból épülnek fel, melyek között koordinatív kovalens kötések jönnek létre. Hosszú ideig azonban komoly kihívást jelentett ezen anyagok tulajdonságainak, különösen színüknek és mágneses viselkedésüknek a megmagyarázása. A 20. század elején megjelenő kristálytér elmélet (Crystal Field Theory, CFT) forradalmasította a komplex vegyületek megértését, egy elegáns és viszonylag egyszerű modell segítségével magyarázva számos megfigyelt jelenséget.
Mielőtt a kristálytér elmélet részleteibe merülnénk, érdemes felidézni, hogy a komplex vegyületek leírására korábban is léteztek elméletek. A valence bond theory (VBT), azaz a vegyértékkötés elmélet például jól magyarázta a kötések irányultságát és a komplexek geometriáját, de nehézségekbe ütközött a szín és a mágneses tulajdonságok kvantitatív értelmezésénél. A kristálytér elmélet éppen ezekre a hiányosságokra kínált megoldást, alapvetően a d-elektronok viselkedésére fókuszálva a ligandumok elektrosztatikus terében. Az elmélet alapvető feltételezése, hogy a fémion és a ligandumok közötti kölcsönhatás tisztán elektrosztatikus jellegű, azaz ionos kötésről van szó, ahol a ligandumokat ponttöltésekként vagy dipólusokként kezeljük, melyek a fémion d-pályáira hatnak.
A kristálytér elmélet történeti háttere és kialakulása
A kristálytér elmélet gyökerei az 1930-as évekbe nyúlnak vissza. Az elméletet elsőként Hans Bethe német fizikus dolgozta ki 1929-ben, elsősorban a kristályokban lévő fémionok spektroszkópiai tulajdonságainak magyarázatára. Bethe munkája kezdetben nem a koordinációs kémiában, hanem a szilárdtestfizikában talált alkalmazásra. Az ő alapvető felismerése az volt, hogy egy fémion d-pályáinak degenerációja (azonos energiájú állapotok) megszűnik, amikor azt egy külső elektrosztatikus térbe helyezzük, például egy kristályrácsban lévő anionok közé.
Később, az 1930-as években, John Hasbrouck Van Vleck amerikai fizikus adaptálta Bethe elméletét a koordinációs vegyületekre. Van Vleck mutatta meg, hogy a ligandumok által létrehozott elektrosztatikus tér hasonlóan befolyásolja a központi fémion d-pályáit, mint a kristályrácsban lévő ionok. Ez a kiterjesztés volt az, ami a kristálytér elméletet a koordinációs kémia egyik alapvető eszközévé tette. Az elmélet kezdetben nem aratott azonnali sikert, mivel a kémikusok a kovalens kötés elméleteihez (mint például a VBT) ragaszkodtak, de az 1950-es évekre, különösen Orgel, Dunitz és Nyholm munkássága révén, széles körben elfogadottá vált, miután számos kísérleti megfigyelést sikeresen magyarázott.
A d-orbitálok viselkedése a ligandumtérben
A kristálytér elmélet megértésének kulcsa a d-orbitálok térbeli elrendezésének és a ligandumok elektrosztatikus terével való kölcsönhatásának ismerete. Egy szabad, gázállapotú fémionban az öt d-orbitál (dxy, dxz, dyz, dx²-y², dz²) azonos energiájú, azaz degenerált állapotban van. Ez azt jelenti, hogy az elektronoknak nincs preferenciájuk egyik d-pálya elfoglalására sem, ha nincsen külső hatás.
Amikor azonban a fémiont ligandumok veszik körül, ezek a ligandumok egy elektrosztatikus teret hoznak létre. A ligandumok negatív töltésűek (anionok) vagy dipólusosak (például vízmolekulák), és a negatív végükkel fordulnak a pozitív töltésű fémion felé. Ez a ligandumtér kölcsönhatásba lép a fémion d-elektronjaival. Mivel az elektronok szintén negatív töltésűek, taszítást tapasztalnak a ligandumoktól. Ez a taszítás nem egyenletes mind az öt d-pályára nézve, mivel azok különböző térbeli orientációval rendelkeznek. Ennek következtében a d-pályák degenerációja megszűnik, és azok különböző energiaszintekre hasadnak fel.
Oktaéderes komplexek: A leggyakoribb elrendezés elemzése
Az oktaéderes geometria a leggyakoribb elrendezés a komplex vegyületekben, ahol hat ligandum veszi körül a központi fémiont. Képzeljünk el egy koordináta-rendszert, ahol a fémion a középpontban van, és a ligandumok az x, -x, y, -y, z, -z tengelyek mentén helyezkednek el. Ebben az esetben a d-pályák közül azok, amelyek direkt módon a tengelyek mentén mutatnak, nagyobb taszítást fognak elszenvedni a ligandumoktól, mint azok, amelyek a tengelyek közötti síkokban helyezkednek el.
Az öt d-pálya közül a dx²-y² és a dz² pályák (együttesen eg halmaz) közvetlenül a tengelyek mentén mutatnak. Ezek a pályák a ligandumok felé irányulnak, ezért a bennük lévő elektronok erősebb elektrosztatikus taszítást szenvednek el, ami megnöveli az energiájukat. Ezzel szemben a dxy, dxz, dyz pályák (együttesen t2g halmaz) a tengelyek közötti síkokban helyezkednek el, így kevésbé vannak kitéve a ligandumok taszító hatásának. Ennek eredményeként az energiájuk csökken.
Ez a felhasadás azt jelenti, hogy az öt degenerált d-pálya két energiaszintre oszlik: egy alacsonyabb energiájú, háromszorosan degenerált t2g szintre és egy magasabb energiájú, kétszeresen degenerált eg szintre. A két energiaszint közötti különbséget nevezzük kristálytér felhasadási energiának, vagy oktaéderes esetben Δo (delta oktaéder). Minél nagyobb a Δo értéke, annál erősebb a ligandumtér.
„Az oktaéderes elrendezésben a d-pályák felhasadása a ligandumok térbeli elhelyezkedésének direkt következménye, mely alapvetően befolyásolja a komplexek elektronikus szerkezetét.”
A Δo értéke kulcsfontosságú a komplexek számos tulajdonságának megértésében, például a színükben és mágneses viselkedésükben. Az elektronok a lehető legalacsonyabb energiájú pályákat foglalják el, de a Δo nagyságától és az elektronok közötti párosítási energiától függően eltérő spin állapotok alakulhatnak ki, amiről később részletesebben is szó lesz.
Tetraéderes és négyzetes planáris komplexek felhasadási mintázatai
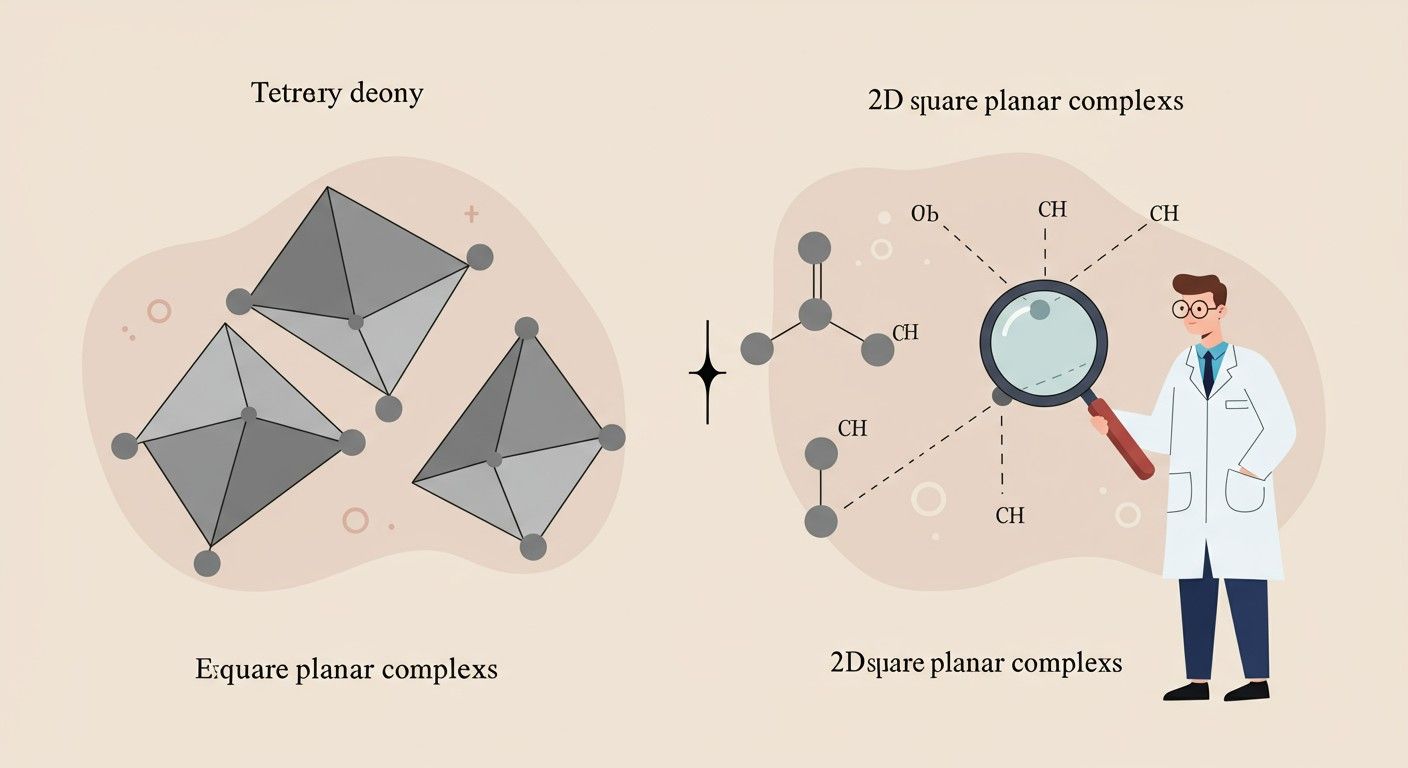
Az oktaéderes mellett más geometriák is előfordulnak a komplex vegyületekben, mint például a tetraéderes és a négyzetes planáris. Ezekben az esetekben a d-orbitálok felhasadási mintázata eltér az oktaéderestől.
Tetraéderes elrendezés: A d-orbitálok felhasadása
Egy tetraéderes komplexben négy ligandum veszi körül a fémiont, és a ligandumok egy tetraéder csúcsaiban helyezkednek el. Képzeljünk el egy kockát, ahol a fémion a középpontban van, és a ligandumok a kocka négy váltakozó sarkánál találhatók. Ebben az elrendezésben a ligandumok nem közvetlenül a tengelyek mentén helyezkednek el, hanem a tengelyek közötti síkok felé mutatnak. Ez azt jelenti, hogy a d-pályák közül a dxy, dxz, dyz (most t2 halmaz, a „g” index hiányzik, mert a tetraéderes környezet nem rendelkezik inverziós centrummal) pályák lesznek közelebb a ligandumokhoz, mint a dx²-y² és a dz² (e halmaz) pályák.
Ennek következtében a tetraéderes ligandumtérben a d-orbitálok felhasadása fordítottja az oktaéderesnek: a t2 pályák energiája emelkedik, míg az e pályák energiája csökken. A felhasadási energiát Δt-vel jelöljük. Fontos megjegyezni, hogy Δt általában sokkal kisebb, mint Δo, tipikusan Δt ≈ (4/9)Δo. Ez a kisebb felhasadási energia azt jelenti, hogy a tetraéderes komplexek szinte mindig magas spinűek.
Négyzetes planáris elrendezés: Az oktaéderes geometriából való származtatás
A négyzetes planáris komplexek tipikusan d8 elektronkonfigurációjú fémionoknál fordulnak elő (pl. Ni2+, Pt2+, Au3+). Ez a geometria úgy képzelhető el, hogy egy oktaéderes komplexből eltávolítjuk a z-tengely mentén lévő két ligandumot. Ennek következtében a d-pályák energiái tovább torzulnak.
A z-tengely menti ligandumok eltávolítása megszünteti a dz² pálya és a ligandumok közötti erős taszítást, így a dz² pálya energiája jelentősen csökken. A dx²-y² pálya, amely a síkban lévő ligandumok felé mutat, a legnagyobb taszítást szenvedi el, így energiája a legmagasabb lesz. A dxy, dxz és dyz pályák energiái is módosulnak. A pontos felhasadási mintázat a következő sorrendet mutatja (növekvő energia szerint): dz² < dxy < dxz/dyz < dx²-y². A dxz és dyz pályák energiája azonos marad a síkban lévő ligandumok irányából nézve.
A négyzetes planáris komplexek felhasadási energiája rendkívül nagy, gyakran nagyobb, mint a legtöbb oktaéderes komplexé, ami a d8 komplexeknél szinte mindig alacsony spinű állapotot eredményez.
A kristálytér felhasadás nagyságát befolyásoló tényezők
A kristálytér felhasadási energia (Δ) nagysága nem állandó, hanem számos tényezőtől függ. Ezek a tényezők magyarázzák, hogy miért olyan sokszínűek a komplex vegyületek tulajdonságai.
A ligandumok természete: A spektrokémiai sor
A legfontosabb tényező a ligandumok természete. Egyes ligandumok erősebb teret hoznak létre, mint mások, ami nagyobb Δ értéket eredményez. Ezt a jelenséget a spektrokémiai sor írja le, amelyben a ligandumok erősségük szerint vannak rendezve, a gyenge tér ligandumoktól az erős tér ligandumokig:
I– < Br– < S2- < SCN– (S-kötés) < Cl– < NO3– < F– < OH– < C2O42- < H2O < NCS– (N-kötés) < py (piridin) < NH3 < en (etilén-diamin) < bipy (bipiridin) < phen (fenantrolin) < NO2– (N-kötés) < PPh3 < CN– < CO
A sor bal oldalán lévő ligandumok (pl. I–, Br–, Cl–) gyenge tér ligandumok, amelyek kis Δ értéket eredményeznek. Ezek jellemzően halogének vagy kén tartalmú ligandumok. A sor jobb oldalán lévő ligandumok (pl. CN–, CO, NH3) erős tér ligandumok, amelyek nagy Δ értéket okoznak. Ezek gyakran nitrogén- vagy szén donor ligandumok. A spektrokémiai sor empirikus sorrend, azaz kísérleti úton határozták meg, és a kristálytér elmélet önmagában nem képes teljes mértékben megmagyarázni (ez a Ligandumtér elmélet egyik fő előnye).
A fémion oxidációs állapota
A központi fémion oxidációs állapota szintén jelentős hatással van a Δ értékére. Minél magasabb a fémion oxidációs állapota, annál nagyobb a pozitív töltése, és annál erősebben vonzza a ligandumokat. Ez közelebb hozza a ligandumokat a fémionhoz, ami erősebb elektrosztatikus kölcsönhatást és nagyobb kristálytér felhasadást eredményez. Például egy Fe3+ komplexben a Δo nagyobb lesz, mint egy Fe2+ komplexben az azonos ligandumok esetén.
A fémion periódusos rendszerbeli helyzete (főkvantumszám)
A fémion periódusos rendszerbeli helyzete, különösen a főkvantumszáma is befolyásolja a Δ értékét. Ugyanazon csoporton belül lefelé haladva (azaz növekvő főkvantumszámmal) a d-orbitálok kiterjedtebbé válnak, ami jobb átfedést és erősebb kölcsönhatást tesz lehetővé a ligandumokkal. Ennek eredményeként a 4d és 5d átmenetifémek komplexei általában sokkal nagyobb Δ értékkel rendelkeznek, mint a 3d átmenetifémek komplexei. Ezért a 4d és 5d fémek komplexei szinte mindig alacsony spinűek, függetlenül a ligandum típusától.
A komplex geometria hatása
Végül, ahogy már említettük, a komplex geometriája is alapvetően meghatározza a d-orbitálok felhasadási mintázatát és a Δ nagyságát. Az oktaéderes, tetraéderes és négyzetes planáris geometriák eltérő felhasadási sémákat mutatnak, és a felhasadási energiák nagyságrendje is eltérő. Δsp > Δo > Δt, ahol Δsp a négyzetes planáris felhasadási energia.
Mágneses tulajdonságok és a spin állapotok
A kristálytér elmélet egyik legfontosabb sikere a komplex vegyületek mágneses tulajdonságainak magyarázata volt. A komplexek mágneses viselkedését a párosítatlan elektronok száma határozza meg. A paramágneses anyagok párosítatlan elektronokkal rendelkeznek, és külső mágneses térbe helyezve vonzódnak hozzá. A diamágneses anyagok minden elektronja párosított, és külső mágneses térben taszítást mutatnak.
Az oktaéderes komplexekben a d-elektronok a felhasadt t2g és eg pályákra kerülnek. A kristálytér felhasadási energia (Δo) és az elektronok párosításához szükséges energia (P, párosítási energia) közötti viszony dönti el, hogy egy komplex magas spinű vagy kis spinű lesz-e.
Nagy spinű és kis spinű komplexek
- Nagy spinű komplexek: Ha a kristálytér felhasadási energia (Δo) kisebb, mint a párosítási energia (P), az elektronok inkább elfoglalják a magasabb energiájú eg pályákat, mielőtt párosítanák magukat a t2g pályákon. Ez maximális számú párosítatlan elektront eredményez, innen a „nagy spinű” elnevezés. Jellemzően gyenge tér ligandumok (pl. H2O, F–) eredményeznek nagy spinű komplexeket.
- Kis spinű komplexek: Ha a kristálytér felhasadási energia (Δo) nagyobb, mint a párosítási energia (P), az elektronok inkább párosodnak az alacsonyabb energiájú t2g pályákon, mielőtt elfoglalnák a magasabb energiájú eg pályákat. Ez minimális számú párosítatlan elektront eredményez, vagy akár teljesen párosított állapotot. Erős tér ligandumok (pl. CN–, CO) jellemzően kis spinű komplexeket hoznak létre.
Ez a jelenség csak a d4-től d7 elektronkonfigurációjú fémionoknál releváns oktaéderes komplexekben, mivel d1, d2, d3 esetén mindig a t2g pályákra kerülnek az elektronok párosítás nélkül, d8, d9, d10 esetén pedig a t2g pályák telítettek, és az eg pályákra kerülnek a további elektronok, így a spin állapot egyértelmű. Tetraéderes komplexeknél a Δt mindig kicsi, ezért szinte kivétel nélkül magas spinűek.
A mágneses momentum számítása
A mágneses momentum (μ) a párosítatlan elektronok számából számítható ki a spin-only képlettel: μ = √n(n+2) Bohr magnetonban (BM), ahol n a párosítatlan elektronok száma. A kísérletileg meghatározott mágneses momentum összehasonlítása az elméleti értékkel lehetővé teszi a spin állapot megállapítását és megerősíti a kristálytér elmélet előrejelzéseit.
A komplexek színe: A kristálytér elmélet magyarázata
Talán a kristálytér elmélet legszenzációsabb sikere a komplex vegyületek élénk és változatos színeinek magyarázata volt. A legtöbb átmenetifém-komplex színes, míg a főcsoportbeli elemek komplexei jellemzően színtelenek. A szín a d-d átmeneteknek köszönhető.
d-d átmenetek és a fényelnyelés
Amikor fehér fény halad át egy átmenetifém-komplex oldaton, a komplex elnyeli a látható fény spektrumának bizonyos hullámhosszait. Az elnyelt energia pontosan megegyezik a Δo (vagy Δt, Δsp) értékével, azaz az alacsonyabb energiájú d-pályáról (t2g) a magasabb energiájú d-pályára (eg) történő elektronátmenethez szükséges energiával. Ezt nevezzük d-d átmenetnek. Azok a hullámhosszak, amelyeket a komplex elnyel, eltűnnek a fehér fényből, és a szemünk az el nem nyelt, áthaladó vagy visszavert fényt látja, mint a komplex színét. Ez a kiegészítő színek elvére épül.
Például, ha egy komplex a kék-zöld fényt nyeli el (kb. 500 nm), akkor a vörös színt látjuk. Ha a vöröset nyeli el (kb. 650 nm), akkor a kék-zöldet látjuk. A spektrokémiai sorban lévő ligandumok különböző Δ értékeket okoznak, így különböző hullámhosszúságú fényt nyelnek el, ami magyarázza a ligandumoktól függő színváltozásokat. Például a [Co(H2O)6]2+ halvány rózsaszín (elnyeli a zöldet), míg a [Co(NH3)6]3+ sárga (elnyeli a kéket), mert az NH3 erősebb tér ligandum, mint a H2O, így nagyobb Δ értéket, és rövidebb hullámhosszú fény elnyelését okozza.
Szelekciós szabályok (Laporte és spin)
A d-d átmenetek azonban nem mindig engedélyezettek. Két fontos szelekciós szabály korlátozza őket:
- Laporte-szabály: Ez a szabály kimondja, hogy az átmenetek, amelyek során a paritás (szimmetria) nem változik, tiltottak. Mivel a d-pályák mind szimmetrikusak az inverziós centrumra nézve (azaz „g” szimmetriájúak), a d-d átmenetek a Laporte-szabály szerint tiltottak lennének. Azonban a komplexekben a vibrációs mozgások (vibronikus csatolás) miatt a szimmetria időlegesen megsérülhet, ami lehetővé teszi az átmeneteket, de gyenge intenzitásúak maradnak.
- Spin-szelekciós szabály: Ez a szabály kimondja, hogy az átmenetek során a spin állapotnak változatlannak kell maradnia (ΔS = 0). Ha egy elektron átmenete során a spin megváltozna, az az átmenet tiltott lenne, vagy rendkívül gyenge intenzitású.
A d-d átmenetek gyenge intenzitása (kis moláris abszorpciós koefficiens) a Laporte-szabály részleges tiltásának köszönhető. Azon komplexek, amelyekben a d-d átmenetek spin-tiltottak is, még halványabb színűek, vagy színtelenek lehetnek.
A spektroszkópia szerepe a kristálytér paraméterek meghatározásában
Az UV-Vis spektroszkópia létfontosságú eszköz a kristálytér paraméterek meghatározásában. A komplexek abszorpciós spektrumának mérésével azonosíthatók a d-d átmenetekhez tartozó abszorpciós sávok, és ezek hullámhosszából (vagy energiájából) közvetlenül meghatározható a Δ értéke. Ez a kísérleti adat kulcsfontosságú az elmélet validálásához és a ligandumok spektrokémiai sorrendjének pontosításához.
Kristálytér stabilizációs energia (CFSE) és termodinamikai stabilitás

A kristálytér stabilizációs energia (Crystal Field Stabilization Energy, CFSE) egy másik alapvető fogalom, amelyet a kristálytér elmélet vezetett be. Ez az energia az az extra stabilitás, amelyet egy fémion szerez, amikor ligandumtérbe kerül, és a d-pályái felhasadnak, összehasonlítva egy hipotetikus, szférikusan szimmetrikus ligandumtérrel (ahol a d-pályák energiája átlagosan megegyezik a felhasadás előtti állapotéval).
A CFSE fogalma és számítása
Egy oktaéderes komplexben a t2g pályák energiája 0.4Δo-val csökken, míg az eg pályák energiája 0.6Δo-val növekszik a barycentrumhoz (az átlagos d-pálya energia) képest. A CFSE-t úgy számítjuk ki, hogy összeadjuk az egyes elektronok stabilizációs járulékát, figyelembe véve a párosítási energiát is, ha az elektronok párosodnak. Például:
- d1: 1 elektron a t2g-n → CFSE = 1 × (-0.4Δo) = -0.4Δo
- d3: 3 elektron a t2g-n → CFSE = 3 × (-0.4Δo) = -1.2Δo
- d8 (oktaéderes): 6 elektron a t2g-n, 2 az eg-n → CFSE = 6 × (-0.4Δo) + 2 × (0.6Δo) = -2.4Δo + 1.2Δo = -1.2Δo
A párosítási energia (P) hozzáadódik a CFSE-hez minden párosított elektronpár után, ami a felhasadás miatt alakul ki. Például egy d4 kis spinű komplexben 4 elektron van a t2g-n, és egy párosítás történt a felhasadás miatt. Ekkor CFSE = 4 × (-0.4Δo) + P = -1.6Δo + P.
A CFSE hatása a komplexek stabilitására és reakciókészségére
A CFSE értéke közvetlenül kapcsolódik a komplex vegyületek termodinamikai stabilitásához. Minél nagyobb a negatív CFSE érték, annál stabilabb a komplex. Ez a stabilitás megnyilvánulhat a képződési entalpiában, a rácsenergiában vagy a hidratációs entalpiában. A CFSE magyarázza a „stabilizációs görbék” jelenségét, ahol bizonyos d-elektron konfigurációjú ionok (pl. d3, d8) stabilabbak, mint a szomszédosak. Ez a stabilitás befolyásolja a fémionok előfordulását és kémiai reakciókészségét is.
„A kristálytér stabilizációs energia segít megérteni, miért bizonyos d-elektron konfigurációjú komplexek kiemelkedően stabilak, és miért mutatnak eltérő reakciókészséget.”
A CFSE-nek köszönhetően egyes fémionok könnyebben képeznek komplexeket, vagy preferálnak bizonyos geometriákat. Például a d8 konfigurációjú Ni2+, Pd2+, Pt2+ ionok hajlamosak négyzetes planáris komplexeket alkotni, mert ez a geometria rendkívül nagy CFSE-t biztosít számukra, különösen erős tér ligandumokkal.
A Jahn-Teller effektus: Geometriai torzulások magyarázata
A kristálytér elmélet egy másik jelentős előrejelzése a Jahn-Teller effektus volt. Ez az effektus egy geometriai torzulást ír le, amely bizonyos d-elektron konfigurációjú komplexekben figyelhető meg, és amely az elektronikus degeneráció megszüntetésére irányul.
Az effektus lényege és okai
A Jahn-Teller tétel kimondja, hogy ha egy nem-lineáris molekulának degenerált elektronikus alapállapota van, akkor a molekula torzulni fog, hogy megszüntesse ezt a degenerációt, és ezzel csökkentse az energiáját. A koordinációs kémiában ez azt jelenti, hogy ha a d-pályákon lévő elektronok aszimmetrikusan töltik be a degenerált eg vagy t2g pályákat (vagyis ha az eg vagy t2g pályák nincsenek teljesen betöltve vagy félig betöltve), akkor a komplex geometriai torzulást szenved el. Ez a torzulás csökkenti az elektronikus energiát, bár a molekula deformációja növeli a nukleáris energiát. Az egyensúlyi állapotban a teljes energia minimalizálódik.
Mikor várható a Jahn-Teller torzulás?
A Jahn-Teller effektus leggyakrabban akkor figyelhető meg, ha az eg pályák aszimmetrikusan vannak betöltve. Ez történik d9 (pl. Cu2+), d4 (magas spinű, pl. Cr2+), és d7 (kis spinű, pl. Co2+) oktaéderes komplexekben. Ilyenkor a torzulás jelentős, és általában a tengelyek mentén lévő ligandumok távolságának változásában (megnyúlás vagy összenyomás) nyilvánul meg. A t2g pályák aszimmetrikus betöltése is okozhat Jahn-Teller torzulást (pl. d1, d2, d6 kis spinű, d7 magas spinű), de ez általában sokkal gyengébb, és nehezebben észlelhető.
Példák Jahn-Teller torzulással rendelkező komplexekre
A Cu2+ (d9) komplexek klasszikus példái a Jahn-Teller torzulásnak. Egy oktaéderes Cu2+ komplexben az eg pályák aszimmetrikusan vannak betöltve (egyik pálya 2, a másik 1 elektronnal). Ennek eredményeként a komplex torzul, általában a z-tengely mentén lévő ligandumok megnyúlásával (tetragonális torzulás). Ez a torzulás megszünteti az eg pályák degenerációját, csökkentve az elektronikus energiát. A Cu2+ ionok jellemzően kék színűek, és a Jahn-Teller torzulás hozzájárul a spektrumukban megfigyelhető széles abszorpciós sávokhoz is.
A kristálytér elmélet alkalmazásai a kémiában és a biológiában
A kristálytér elmélet nem csupán elméleti érdekesség, hanem széles körű gyakorlati alkalmazásokkal is rendelkezik, mind a kémia, mind a biológia területén.
Katalízis és koordinációs polimerek
Az átmenetifém-komplexek kiváló katalizátorok számos ipari folyamatban, például polimerizációs reakciókban, hidrogénezésben vagy oxidációban. A katalizátorok aktivitása és szelektivitása szorosan összefügg a fémion elektronikus szerkezetével és a ligandumok által létrehozott ligandumtérrel. A kristálytér elmélet segít megérteni, hogyan befolyásolja a ligandumok jellege a fémion d-pályáinak energiáját, ami kulcsfontosságú a reakció mechanizmusainak és az átmeneti állapotok stabilitásának megértéséhez. A CFSE például befolyásolja a ligandumcserék sebességét és a redoxpotenciálokat.
A koordinációs polimerek vagy fém-organikus vázanyagok (MOF-ok) szintén profitálnak a kristálytér elmélet megértéséből. Ezek a porózus anyagok nagy felülettel rendelkeznek, és gáztárolásra, elválasztásra vagy katalízisre használhatók. A fémionok és a szerves ligandumok közötti kölcsönhatások megértése elengedhetetlen az ilyen anyagok tervezéséhez és tulajdonságaik finomhangolásához.
Biokémiai rendszerek: Hemoglobin, klorofill és fémionok szerepe
A biológiai rendszerekben számos alapvető folyamatban szerepet játszanak az átmenetifém-komplexek. A hemoglobin, amely az oxigént szállítja a vérben, egy vas(II) iont tartalmazó porfirin komplex. A vas(II) ion d6 konfigurációjú, és a ligandumtér befolyásolja a spin állapotát. Az oxigénkötés során a vas(II) ion spin állapota megváltozik (magas spinűből kis spinűvé), ami a porfirin gyűrű konformációjának változásához vezet, és ezáltal az oxigénkötés affinitását szabályozza. A kristálytér elmélet segít megmagyarázni ezeket az elektronikus változásokat és a hemoglobin működésének alapjait.
Hasonlóképpen, a klorofill, amely a fotoszintézisben kulcsszerepet játszik, egy magnézium(II) iont tartalmazó porfirin komplex. Bár a magnézium nem átmenetifém, a kristálytér elmélet elvei általánosabban is alkalmazhatók a fémionok és a ligandumok közötti kölcsönhatások megértésére az elektronikus szerkezet szempontjából. Más fémionok, mint például a kobalt a B12-vitaminban, vagy a cink számos enzimben, szintén létfontosságú szerepet töltenek be, és a kristálytér elmélet alapelvei segítenek megérteni funkciójukat.
Anyagtudomány: Pigmentek, mágneses anyagok
Az anyagtudományban is széles körben alkalmazzák a kristálytér elméletet. A pigmentek és színezékek színét gyakran átmenetifém-komplexek adják, és a kristálytér felhasadás magyarázza a színüket. A különböző fémionok és ligandumok kiválasztásával széles színskála hozható létre.
A mágneses anyagok fejlesztésében is kulcsfontosságú a kristálytér elmélet. Az átmenetifém-ionok mágneses tulajdonságai, mint például a paramágnesesség vagy a ferromágnesesség, közvetlenül kapcsolódnak a párosítatlan d-elektronok számához és elrendeződéséhez. Az elmélet segít megtervezni olyan anyagokat, amelyek specifikus mágneses tulajdonságokkal rendelkeznek, például mágneses adathordozókhoz vagy spintronikai alkalmazásokhoz.
A kristálytér elmélet korlátai és a Ligandumtér elmélet
Bár a kristálytér elmélet rendkívül sikeresen magyarázott számos jelenséget, mint minden modellnek, ennek is vannak korlátai. A főbb hiányosságok a következők:
A tisztán ionos modell hiányosságai
A kristálytér elmélet alapvető feltételezése, hogy a fémion és a ligandumok közötti kötés tisztán ionos jellegű, azaz elektrosztatikus vonzásról van szó. Ez a modell azonban nem veszi figyelembe a kovalens kötés bizonyos mértékű jelenlétét, amely valójában minden koordinációs vegyületben megfigyelhető. A kovalens jelleg figyelmen kívül hagyása problémákat okoz a spektrokémiai sor teljes magyarázatában és a ligandumok egymás közötti különbségeinek részletesebb értelmezésében.
A kovalens jellegek figyelmen kívül hagyása
A kristálytér elmélet nem tudja megmagyarázni a π-kötések szerepét, amelyek bizonyos ligandumok (pl. CO, CN–, NO) és a fémion között kialakulhatnak. Ezek a π-kötések jelentősen befolyásolják a kristálytér felhasadás nagyságát, és az elmélet nem képes erre magyarázatot adni, mivel csak a σ-kölcsönhatásokat (vagyis az elektrosztatikus taszítást) veszi figyelembe.
A spektrokémiai sor magyarázatának nehézségei
Ahogy már említettük, a spektrokémiai sor egy empirikus sorrend. Bár a kristálytér elmélet megmagyarázza, hogy a ligandumok erőssége befolyásolja a Δ értékét, nem képes előre jelezni, hogy miért van egy ligandum (pl. CN–) sokkal erősebb tér ligandum, mint egy másik (pl. F–), tisztán elektrosztatikus alapon. A fluorid ion például erősen negatív töltésű, és az elmélet szerint erős tér ligandumnak kellene lennie, de valójában gyenge tér ligandum.
A Ligandumtér elmélet mint továbbfejlesztés
A kristálytér elmélet korlátainak leküzdésére fejlesztették ki a Ligandumtér elméletet (Ligand Field Theory, LFT). Az LFT a kristálytér elméletet és a molekulaorbitál elméletet (Molecular Orbital Theory, MOT) ötvözi. Az LFT figyelembe veszi a fémion és a ligandumok közötti kovalens kölcsönhatásokat is, beleértve a σ- és π-kötéseket. Ezáltal az LFT sokkal pontosabb és átfogóbb képet ad a komplex vegyületek elektronikus szerkezetéről és tulajdonságairól, képes megmagyarázni a spektrokémiai sort és a kovalens jellegek hatását.
A kristálytér elmélet és a molekulaorbitál elmélet kapcsolata

A kristálytér elmélet és a molekulaorbitál elmélet (MOT) két különböző megközelítést kínál a kémiai kötések leírására, de a koordinációs kémiában gyakran kiegészítik egymást. Míg a CFT viszonylag egyszerű, elektrosztatikus modellen alapul, addig a MOT sokkal kifinomultabb, kvantummechanikai alapon írja le az atomi pályák kombinációjából kialakuló molekulaorbitálokat.
A két elmélet közötti átfedések és különbségek
A CFT fő előnye az egyszerűsége és intuitív jellege. Könnyen alkalmazható a d-orbitálok felhasadásának és a komplexek alapvető tulajdonságainak (szín, mágnesesség) magyarázatára anélkül, hogy bonyolult kvantummechanikai számításokra lenne szükség. A CFT azonban elhanyagolja a kovalens jelleget, ami korlátozza a pontosságát és magyarázó erejét.
A MOT ezzel szemben a kovalens kötéseket is figyelembe veszi, és pontosabb leírást ad a fém-ligandum kötésekről, beleértve a σ- és π-kötéseket is. A MOT segítségével a spektrokémiai sor is kvantitatívan magyarázható, mivel figyelembe veszi a ligandumok π-donor vagy π-akceptor képességét. A MOT szerint az atomi pályák (fém d-pályák és ligandum pályák) kombinálódnak, hogy kötő, nemkötő és lazító molekulaorbitálokat hozzanak létre. A d-pályák felhasadása a MOT keretében a fém d-pályái és a ligandum pályái közötti kölcsönhatás eredményeként értelmezhető.
Hogyan egészítik ki egymást a komplex vegyületek leírásában?
A két elmélet nem egymás kizárója, hanem inkább kiegészítik egymást. A kristálytér elmélet továbbra is hasznos eszköz a komplex vegyületek kezdeti megértéséhez és a kvalitatív előrejelzésekhez. Gyakran használják pedagógiai eszközként is, hogy bevezessék a hallgatókat a koordinációs kémia alapjaiba. Amikor azonban mélyebb, kvantitatívabb elemzésre van szükség, vagy amikor a kovalens jellegek dominálnak, a molekulaorbitál elmélet (vagy annak kiterjesztése, a Ligandumtér elmélet) nyújt pontosabb leírást.
A Ligandumtér elmélet tulajdonképpen a CFT és a MOT egyfajta szintézise. A Ligandumtér elmélet megtartja a CFT-ből a felhasadt d-pályák fogalmát, de a felhasadást már nem tisztán elektrosztatikus, hanem a molekulaorbitálok képződéséből eredő jelenségként magyarázza. Ez a hibrid megközelítés teszi lehetővé a komplex vegyületek tulajdonságainak rendkívül pontos és átfogó leírását.
Fejlett koncepciók és további megfontolások
A kristálytér elmélet alapjainak megértése után érdemes megemlíteni néhány fejlettebb koncepciót, amelyek tovább finomítják a komplex vegyületek elektronikus szerkezetének leírását.
Nephelauxetikus effektus
A nephelauxetikus effektus (görögül „felhő-tágulás”) arra utal, hogy a d-elektronok a ligandumtérben kevésbé lokalizáltak a fémionon, mint egy szabad fémionban. Ez azt jelenti, hogy a d-orbitálok kiterjedtebbé válnak, és a ligandumokkal való kovalens kölcsönhatás miatt csökken a taszítás az elektronok között. Ennek következtében a párosítási energia (P) és a Δ értéke is csökken. A nephelauxetikus sorrend a ligandumok azon képességét írja le, hogy mennyire képesek csökkenteni a fémion d-elektronjainak taszítását, azaz mennyire teszik „felhőssé” a d-pályákat. Ez a sorrend is empirikus, és a kovalens jellegek meglétére utal, ami a kristálytér elmélet tisztán ionos modelljével ellentétes.
Spin-crossover komplexek
Néhány komplex vegyületben, különösen a d4-d7 konfigurációjú fémionok esetén, a Δ és a párosítási energia (P) értéke nagyon közel áll egymáshoz. Ilyen esetekben a komplex képes spin-crossover jelenséget mutatni, azaz hőmérséklet, nyomás vagy fény hatására átmehet magas spinű állapotból kis spinű állapotba, és fordítva. Ez a jelenség jelentős változásokkal jár a mágneses, optikai és szerkezeti tulajdonságokban, és potenciális alkalmazásokat kínál a molekuláris kapcsolókban vagy adathordozókban.
Relativisztikus effektusok a nehézfémek komplexekben
A nehézfémek (pl. 5d átmenetifémek) komplex vegyületeiben a relativisztikus effektusok is jelentős szerepet játszanak. Ezek az effektusok a gyorsan mozgó elektronok viselkedéséből adódnak, és befolyásolják az orbitálok energiáját és kiterjedését. A relativisztikus hatások például hozzájárulnak a 5d fémek kiemelkedően nagy kristálytér felhasadásához és különleges kémiai tulajdonságaikhoz, amelyek eltérnek a 3d és 4d analógokétól. Bár a kristálytér elmélet önmagában nem tartalmazza ezeket a komplex hatásokat, a modern elméleti modellek figyelembe veszik őket a pontosabb előrejelzések érdekében.
A kristálytér elmélet öröksége és jövőbeli perspektívái
A kristálytér elmélet, annak ellenére, hogy több mint 90 éve született, továbbra is alapvető sarokköve a koordinációs kémia oktatásának és kutatásának. Egyszerűsége és intuitív jellege miatt kiváló bevezetőt nyújt a komplex vegyületek elektronikus szerkezetének megértéséhez. Bár a modern kvantumkémiai módszerek és a Ligandumtér elmélet pontosabb leírást adnak, a CFT által bevezetett fogalmak – mint a d-orbitálok felhasadása, a Δ érték, a magas/kis spin állapotok, a CFSE, a Jahn-Teller effektus – továbbra is kulcsfontosságúak a területen.
Az elmélet tartós hatása abban rejlik, hogy egy keretrendszert biztosított, amely lehetővé tette a kémikusok számára, hogy rendszerezzenek és megmagyarázzanak számos megfigyelt jelenséget, mint például a komplexek színét, mágneses tulajdonságait és stabilitását. Ez a keretrendszer szolgált alapul a későbbi, kifinomultabb elméletek, mint a Ligandumtér elmélet és a molekulaorbitál elmélet fejlesztéséhez is. A CFT egyszerűsége segített abban, hogy a koordinációs kémia fogalmai szélesebb körben elterjedjenek és megérthetővé váljanak.
A modern számítási módszerek, mint például a sűrűségfunkcionál-elmélet (DFT), ma már lehetővé teszik a komplex vegyületek elektronikus szerkezetének és tulajdonságainak rendkívül pontos előrejelzését. Ezek a módszerek azonban gyakran a kristálytér elmélet által bevezetett alapfogalmakat használják referenciapontként, vagy az azokból származó paramétereket finomítják. Az elmélet relevanciája a mai kutatásokban is megmarad, különösen az új koordinációs vegyületek tervezésénél, a katalizátorok optimalizálásánál, és a biológiai rendszerekben szerepet játszó fémionok funkciójának feltárásánál. A kristálytér elmélet tehát nem csupán egy történelmi mérföldkő, hanem egy élő és fejlődő koncepció, amely továbbra is inspirálja a kémikusokat a komplex vegyületek lenyűgöző világának felfedezésére.







